
Abstract
Background
Competing endogenous RNA (ceRNA) represents a class of RNAs (e.g., long noncoding RNAs [lncRNAs]) with microRNA (miRNA) binding sites, which can competitively bind miRNA and inhibit its regulation of target genes. Increasing evidence has underscored the involvement of dysregulated ceRNA networks in the occurrence and progression of colorectal cancer (CRC). The purpose of this study was to construct a ceRNA network related to the prognosis of CRC and further explore the potential mechanisms that affect this prognosis.
Methods
RNA-Seq and miRNA-Seq data from The Cancer Genome Atlas (TCGA) were used to identify differentially expressed lncRNAs (DElncRNAs), microRNAs (DEmiRNAs), and mRNAs (DEmRNAs), and a prognosis-related ceRNA network was constructed based on DElncRNA survival analysis. Subsequently, pathway enrichment, Pearson correlation, and Gene Set Enrichment Analysis (GSEA) were performed to determine the function of the genes in the ceRNA network. Gene Expression Profiling Interactive Analysis (GEPIA) and immunohistochemistry (IHC) were also used to validate differential gene expression. Finally, the correlation between lncRNA and immune cell infiltration in the tumor microenvironment was evaluated based on the CIBERSORT algorithm.
Results
A prognostic ceRNA network was constructed with eleven key survival-related DElncRNAs (MIR4435-2HG, NKILA, AFAP1-AS1, ELFN1-AS1, AC005520.2, AC245884.8, AL354836.1, AL355987.4, AL591845.1, LINC02038, and AC104823.1), 54 DEmiRNAs, and 308 DEmRNAs. The MIR4435-2HG- and ELFN1-AS1-associated ceRNA subnetworks affected and regulated the expression of the COL5A2, LOX, OSBPL3, PLAU, VCAN, SRM, and E2F1 target genes and were found to be related to prognosis and tumor-infiltrating immune cell types.
Conclusions
MIR4435-2HG and ELFN1-AS1 are associated with prognosis and tumor-infiltrating immune cell types and could represent potential prognostic biomarkers or therapeutic targets in colorectal carcinoma.
Similar content being viewed by others


Background
Colorectal cancer (CRC), the third most common cancer and the second leading cause of cancer mortality worldwide, is caused by the accumulation of long-term genetic mutations and epigenetic alterations [1]. Although recent interventional and therapeutic advances have greatly improved the overall survival (OS) of CRC, patients with advanced CRC still show poor prognosis due to the high frequency of recurrence and metastasis [2]. Investigation of the molecular mechanisms underlying CRC, from initiation to metastasis, is urgently needed to improve its early diagnosis, treatment, and prognosis. Long noncoding RNAs (lncRNAs) are noncoding transcripts that range from 200 to 100,000 nucleotides in length and lack protein-coding capacity. They function as signals, decoys, guides, and scaffolds in gene regulatory networks [3]. The regulatory mechanism of lncRNA depends on its subcellular localization. Cytoplasmic lncRNAs primarily participate in post-transcriptional and translational regulation by acting as microRNA (miRNA) sponges and modulating mRNA stability. An increasing number of lncRNAs have been found to participate in CRC initiation and progression by functioning as competing endogenous RNAs (ceRNAs) [4,5,6]. Specifically, lncRNAs derepress target gene expression by competitively binding to shared sequences of miRNAs [7, 8]. Recent studies showed that ceRNA crosstalk was involved in various biological processes, including CRC cell proliferation, invasion, epithelial-to-mesenchymal transition (EMT), metastasis, and immune infiltration [9,10,11,12,13].
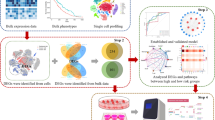
In the present study, we acquired the expression profiles and clinical information for colon adenocarcinoma (COAD) and rectal adenocarcinoma (READ) from The Cancer Genome Atlas (TCGA). Differentially expressed mRNAs (DEmRNAs), lncRNAs (DElncRNAs), and miRNAs (DEmiRNAs) were then identified by comparing tumor samples and normal samples. We performed survival analysis and predicted RNA interaction pairs to construct a prognosis-related ceRNA network. The protein–protein interaction (PPI) network was embodied in Cytoscape to identify module clusters and hub genes. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were performed to explore the biological functions of the mRNAs in the ceRNA and module network. In addition, we analyzed the correlations between key lncRNAs and the clinical features of CRC patients. Co-expression analysis between lncRNAs and target genes was performed to verify the ceRNA hypothesis. Eventually, we established ceRNA regulatory subnetworks based on the above results. GEPIA and immunohistochemistry were used to validate the differential expression of genes, whereas GSEA was used to predict the underlying functions of the genes in the subnetwork. The CIBERSORT algorithm was used to estimate the proportion of immune cells for further correlation analysis (Fig. 1).

Flowchart for the identification and analysis of prognostic lncRNAs in the ceRNA network
Methods
Data downloading and pre-processing
The mRNA, lncRNA, and miRNA expression profiles of the CRC patients were downloaded from TCGA database (https://cancergenome.nih.gov/). We collected RNA-Seq data for 612 samples (568 CRC and 44 normal samples) in HTSeq-Counts files and miRNA-Seq data for 548 samples (539 CRC and nine normal samples). The sequence data were derived from Illumina HiSeq RNASeq and miRNASeq platforms, which were publicly available. GENCODE (https://www.gencodegenes.org/) was used to convert the RNA-Seq data into lncRNAs (sense_overlapping, lincRNA, 3prime_overlapping_ncRNA, processed_transcript, antisense, and sense_intronic) and mRNAs (protein-coding).
Differential expression analysis of RNAs
To screen differentially expressed RNAs (DERNAs) between tumor samples and normal samples, we used the “edgeR” package in R software to standardize and analyze the downloaded data. DEmRNAs, DElncRNAs, and DEmiRNAs were identified with the criteria of |log2 fold change (FC)| > 1.0 and adjusted P value < 0.05. Heatmap clustering and volcano plot were generated using R package “edgeR” to reveal the differential expressions of the DEmRNAs, DElncRNAs, and DEmiRNAs.
Prediction of RNA interaction pairs
The online tool DIANA-LncBase version 3 (http://www.microrna.gr/LncBase) [14] was used to predict the potential miRNAs targeted by DElncRNAs and determine lncRNA–miRNA interaction pairs. MiRDB (http://www.mirdb.org/) [15], miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/) [16], and TargetScan (http://www.targetscan.org/) [17] databases were used to predict the target genes of DEmiRNAs and miRNA–mRNA interaction pairs. The R package “VennDiagram” was used to compare the target genes with the DEmRNAs to obtain overlapping genes.
Survival analysis and ceRNA network construction
RNA expression profiles and related clinical information of the CRC patients were acquired from TCGA database. To identify lncRNAs significantly associated with patient prognosis, we used the R package “survival” to perform Kaplan-Meier curve analysis of DElncRNAs. DElncRNAs with P < 0.05 were considered to be key lncRNAs for construction of the next network. Based on the hypothesis that lncRNA acts as a miRNA sponge to regulate mRNA expression [7], we selected key lncRNAs and lncRNA–miRNA–mRNA interaction pairs to build the ceRNA regulatory network. Ultimately, we combined specific lncRNA–miRNA and miRNA–mRNA pairs to establish a prognosis-related network. The established ceRNA network was visualized using Cytoscape v3.6.0 software [18]. Univariate and multivariate Cox regression analyses were performed to identify the independent prognostic indicators for overall survival (OS).
Construction of PPI network
To better understand the interactions of DEmRNAs in the ceRNA network, we constructed a PPI network using the Search Tool for the Retrieval of Interacting Genes (STRING; https://string-db.org/) [19] database. The median confidence score (0.4) was used to filter protein interactions. Cytoscape 3.6.0 was used to embody the constructed PPI networks. In addition, we used the plugin cytoHubba to identify hub genes in the PPI network according to degree of connectivity. The top 30 hub genes were identified through the Degree ranking method. The MCODE plugin was used to analyze the PPI network and select the top module for subnetwork analysis.
Functional annotation analysis
To explore the potential biological functions and pathways of mRNAs in the ceRNA and module network, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis through the R package “ClusterProfiler”. GO terms and KEGG pathways with P < 0.05 were considered significantly enriched.
Clinical feature analysis of key lncRNAs
Clinical information, including age, sex, tumor stage, and TNM stage, was extracted from TCGA database. Based on the prognosis-related ceRNA network, we evaluated the associations between key lncRNAs and the clinical features of CRC patients using the R package “ggpubr”.
Correlation analysis of lncRNAs and mRNAs
We performed Pearson correlation analysis and selected the positive correlations between the lncRNAs and mRNAs in ceRNA network. Pearson correlation coefficient > 0.4 and P < 0.001 were used as inclusion criteria. Based on the above co-expression relationships, we constructed a ceRNA subnetwork to investigate the potential mechanisms.
Validation of gene expression
Gene Expression Profiling Interactive Analysis (GEPIA; http://gepia.cancer-pku.cn/) is an online analysis tool based on TCGA and the Genotype-Tissue Expression (GTEx) dataset [20]. Differential expression boxplot in GEPIA was used to compare the expression levels of genes between tumor samples and normal samples. The Human Protein Atlas (THPA; https://www.proteinatlas.org) is a database that provides immunohistochemistry-based maps of human protein profiles for cancer tissues and normal tissues and cells [21]. We obtained immunohistochemistry images and data from this database to validate gene expression at the protein level. Staining intensity was evaluated as follows: no staining, 0; low staining, 1; medium staining, 2; high staining, 3. Staining quantity was scored as follows: none, 0; < 25%, 1; 25–50%, 2; 51–75%, 3; > 75%, 4. The total staining score was calculated by multiplying these two scores. Differences in the total staining scores between normal and CRC tissues were evaluated using the unpaired t-test. P < 0.05 was considered statistically significant.
Gene set enrichment analysis
To explore the biological pathways of genes in a subnetwork, Gene Set Enrichment Analysis (GSEA) analysis was performed based on TCGA dataset. According to the cutoff value, the samples were divided into high and low expression phenotypes. The gene set permutations were performed 1000 times per analysis. Gene sets with |normalized enrichment score (NES)| > 1 and P < 0.05 were considered significantly enriched.
Evaluation of immune cell infiltration
Normalized gene expression data were uploaded to TIMER2.0 (http://timer.cistrome.org/) [22], a website that provides comprehensive evaluation and analysis of tumor-infiltrating immune cells, to estimate the relative abundance of 22 immune cell types based on the CIBERSORT algorithm. To evaluate the correlation between lncRNA and immune cell infiltration in the tumor microenvironment, CRC samples from the TCGA database were divided into high and low lncRNA expression groups. The Wilcoxon rank-sum test was performed to compare the differences in immune cell infiltration between the high and low lncRNA expression groups.
Results
Differentially expressed lncRNAs, miRNAs, and mRNAs
We obtained the expression profiles of 19,645 mRNAs, 14,166 lncRNAs, and 1881 miRNAs from TCGA database. A total of 2480 DEmRNAs, 217 DElncRNAs, and 376 DEmiRNAs were identified with the criteria of |log2 FC| > 1 and adjusted P < 0.05. Among them, 1325 DEmRNAs, 159 DElncRNAs, and 243 DEmiRNAs were upregulated, whereas 1155 DEmRNAs, 58 DElncRNAs, and 133 DEmiRNAs were downregulated (Supplementary Figure S1 and Fig. 2).

Volcano plots of differentially expressed RNAs. a Volcano plot of lncRNAs. b Volcano plot of miRNAs. c Volcano plot of mRNAs. The red dots represent upregulated RNAs, and the green dots represent downregulated RNAs
Prediction of lncRNA–miRNA and miRNA–mRNA interactions
We predicted lncRNA–miRNA interaction pairs based on DElncRNAs using the online tool DIANA-LncBase version 3 and found that 153 of the 217 DElncRNAs might target 199 of the 376 DEmiRNAs. Subsequently, miRTarBase, miRDB, and TargetScan databases were used to identify miRNA–mRNA interaction pairs. Target mRNAs that were recognized by all three databases were selected as candidate genes. The results showed that 184 of the 199 specific DEmiRNAs might target 3475 candidate genes. After comparing the 3475 candidate genes with 2480 DEmRNAs, we obtained 455 overlapping genes and 1148 miRNA–mRNA interaction pairs.
Construction of prognosis-related ceRNA network
A ceRNA network was constructed using 2489 lncRNA-miRNA pairs and 1148 miRNA-mRNA pairs using Cytoscape software. The association of 153 lncRNAs in the ceRNA network with patient prognosis was analyzed using Kaplan-Meier survival curves. The results showed that 11 of the 153 lncRNAs were significantly associated with OS. The 11 prognosis-related DElncRNAs were considered key lncRNAs. LINC02038 and AC104823.1 were positively correlated with OS, whereas MIR4435-2HG, NKILA, AFAP1-AS1, ELFN1-AS1, AC005520.2, AC245884.8, AL354836.1, AL355987.4, and AL591845.1 were negatively correlated with OS (Fig. 3). Ultimately, we constructed a survival-related ceRNA network with 11 key lncRNAs, 54 DEmiRNAs, and 308 DEmRNAs. This network contained 373 nodes and 739 edges (Supplementary Figure S2). A total of 11 lncRNAs and 308 mRNAs in the ceRNA network were included in the univariate cox regression analysis. Six lncRNAs and 28 mRNAs with P < 0.05 were subjected to multivariate COX regression analysis. The lncRNA ELFN1-AS1 and the SPTBN2, CCNF, and VEGFA mRNAs were identified as independent prognostic factors for OS in CRC patients (Supplementary Table S3).

Kaplan-Meier survival curves of eleven key lncRNAs related to overall survival (OS). LINC02038 (a) and AC104823.1 (b) were positively correlated with OS. MIR4435-2HG (c), ELFN1-AS1 (d), NKILA (e), AFAP1-AS1 (f), AL591845.1 (g), AL355987.4 (h), AL354836.1 (i), AC005520.2 (j), and AC245884.8 (k) were negatively correlated with OS. The red lines represent the high expression groups, and the blue lines represent the low expression groups
PPI network and module analysis
The mRNAs in the ceRNA network were used to construct a PPI network based on the STRING database. The PPI network was visualized by Cytoscape and included 265 nodes and 1109 edges (Fig. 4). The cytoHubba plugin was used to identify the top 30 hub genes with the highest degrees (Table 1), of which 7 genes (VEGFA, BIRC5, CDC25A, KPNA2, DLGAP5, CCNE1, and CCNF) were significantly related to OS using Kaplan-Meier analysis (Fig. 5). The MCODE plugin was used in the PPI network to identify the top module, which contained 31 nodes and 341 PPIs (Fig. 6a).

The protein–protein interaction (PPI) network of differentially expressed genes in the prognosis-related ceRNA network. Each node corresponds to a protein-coding gene, and the size of the node represents the connection degree

Kaplan-Meier survival curves of seven hub genes related to overall survival (OS). CDC25A (a), BIRC5 (b), CCNE1 (c), CCNF (d), DLGAP5 (e), and KPNA2 (f) were positively correlated with OS, and VEGFA (g) was negatively correlated with OS. The red lines represent the high expression groups, and the blue lines represent the low expression groups

The identification and analysis of the module network. a The top modules selected from the PPI network using the plugin MCODE. b GO enrichment analysis of genes in the module network. Genes are shown on the left side of the chord diagram and enriched GO terms are shown on the right side. Red bars represent upregulated genes and blue bars represent downregulated genes
GO and KEGG enrichment analysis of DEmRNAs in ceRNA
We performed GO and KEGG enrichment analysis on 308 mRNAs in the ceRNA network and 31 mRNAs in the module network with the R package “ClusterProfiler”. GO functional enrichment analysis suggested that mRNAs in the ceRNA network were significantly enriched in “proximal promoter sequence-specific DNA binding”, “transcription coregulator activity”, and “RNA polymerase II proximal promoter sequence-specific DNA binding” (Fig. 7a). KEGG pathway analysis showed that ceRNA-related mRNAs were enriched in “microRNAs in cancer”, “cell cycle”, and “PI3K-Akt signaling pathway” (Fig. 7b). Enrichment analysis of mRNAs in the module network showed that the enriched GO terms included “microtubule binding”, “cyclin-dependent protein serine/threonine kinase regulator activity”, and “tubulin binding”, whereas the most enriched KEGG pathways were “cell cycle”, “cellular senescence”, and “p53 signaling pathway” (Fig. 6b and Fig. 7c).

GO and KEGG enrichment analysis of genes in the ceRNA and module networks. a GO enrichment analysis of genes in the ceRNA network. b KEGG enrichment analysis of genes in the ceRNA network. c KEGG enrichment analysis of genes in the module network
Clinical feature analysis of key lncRNAs
To further investigate the 11 prognosis-related lncRNAs in the ceRNA network, we analyzed the correlation between lncRNAs and clinical features, including age, sex, tumor stage, and TNM stage. Seven lncRNAs were associated with the clinical features of CRC patients (all P < 0.05). Four lncRNAs—NKILA, AC005520.2, AC245884.8, and AL354836.1—were age-related, four lncRNAs—MIR4435-2HG, NKILA, AL591845.1, and AC005520.2—were associated with tumor stage, and six lncRNAs—MIR4435-2HG, ELFN1-AS1, NKILA, AL591845.1, AC005520.2, and AL354836.1—were associated with TNM stage (Fig. 8). The detailed clinical and pathological characteristics of 548 colorectal cancer patients are shown in Table 2.

Clinical feature analysis of eleven key lncRNAs. a Correlations of lncRNA expression with age. b Correlations of lncRNA expression with tumor stage. c Correlations of lncRNA expression with pathologic N stage. d Correlations of lncRNA expression with pathologic M stage
Co-expression analysis of lncRNAs and mRNAs
To verify the hypothesis that lncRNA positively regulates mRNA expression by interacting with miRNA in the ceRNA network, we performed Pearson correlation analysis between seven clinically relevant lncRNAs and their target mRNAs. The results showed that MIR4435-2HG was positively correlated with 15 mRNAs (r > 0.4, Table 3), of which COL5A2, LOX, OSBPL3, PLAU, and VCAN had a significantly positive correlation with MIR4435-2HG (r > 0.5). We speculated that MIR4435-2HG upregulated the expression of the above five DEmRNAs through different DEmiRNAs (mir-29a, mir-424, mir-335, mir-193a, and mir-193a) (Fig. 9a-f). Our correlation analysis of ELFN1-AS1 showed that seven mRNAs were positively associated with ELFN1-AS1 (r > 0.4, Table 3), whereas SRM and E2F1 had a significantly positive correlation with ELFN1-AS1 (r > 0.5). Based on the ceRNA hypothesis, ELFN1-AS1 may upregulate the expression of SRM and E2F1 by acting as sponges of mir-423 and mir-93, respectively (Fig. 9g-i).

Pearson correlation analysis between lncRNAs and mRNAs. a–e The co-expression relationship between MIR4435-2HG and paired mRNAs (PLAU, LOX, COL5A1, VCAN and OSBPL3). f The ceRNA regulatory subnetwork of MIR4435-2HG. (G–H) The co-expression relationship between ELFN1-AS1 and paired mRNAs (SRM and E2F1). i The ceRNA regulatory subnetwork of ELFN1-AS1
Validation of gene expression by GEPIA and immunohistochemistry
We used TCGA and GTEx datasets in GEPIA to further validate the differential expression of genes between tumor and normal samples. The expression levels of PLAU, OSBPL3, LOX, VCAN, E2F1, and SRM were upregulated in COAD and READ tissues compared with normal tissues (all P < 0.05, Fig. 10). However, the expression of COL5A2 showed no differential expression between tumor and normal tissues. Immunohistochemistry results were obtained from the THPA database, and the staining scores were quantified to verify the protein expression levels of the genes. The staining scores for SRM (P = 0.0015), OSBPL3 (P = 0.0263), and VCAN (P = 0.0229) were higher in the CRC tissue than normal tissue, whereas PLAU (P = 0.0169) showed the opposite pattern. There was no significant difference between the E2F1 staining scores (P = 0.6213) for CRC and normal tissue (Fig. 11).

Validation of differential gene expression using Gene Expression Profiling Interactive Analysis (GEPIA). The red bars represent tumor samples, and the gray bars represent normal samples

Immunohistochemical analysis of protein expression levels of genes based on The Human Protein Atlas (THPA) database. a Immunohistochemical results for normal tissues. b Immunohistochemical results for CRC tissues. c Plot diagram of staining scores between CRC and normal tissues
GSEA-predicted biological functions of genes
We used GSEA analysis to predict the enriched KEGG pathways between high and low expressions of genes in a subnetwork. PLAU, encoding a secreted serine protease, was positively correlated with “extracellular matrix”, “angiogenesis”, and “serine hydrolase activity”. LOX was significantly enriched in “extracellular matrix”, “cell adhesion molecule binding”, and “angiogenesis”. VCAN belongs to the aggrecan/versican proteoglycan family, which was positively correlated with “extracellular matrix component”, “cell adhesion molecule binding”, and “proteoglycan metabolic process”. COL5A2 encodes one alpha chain of fibrillar collagen trimers. The high expression of COL5A2 was correlated with “extracellular matrix”, “collagen trimer”, and “collagen fibril organization”, whereas E2F1 was negatively correlated with “biological adhesion”, “regulation of cell adhesion”, and “cell motility” (Fig. 12).

Gene set enrichment analysis (GSEA) revealed enriched KEGG pathways based on gene expression levels
Tumor-infiltrating immune cells in CRC
A cohort of 568 CRC patients was divided into high and low expression groups based on lncRNA expression. The results of the Wilcoxon rank-sum test revealed that plasma B cells (P = 0.001) and CD4+ memory resting T cells (P < 0.001) were greater in patients with low MIR4435-2HG expression, while follicular helper T cells (P = 0.037) and neutrophils (P < 0.001) were greater in patients with high MIR4435-2HG expression (Fig. 13a). The high ELFN1-AS1 expression group had significantly higher numbers of plasma B cells (P = 0.039), CD4+ memory-activated T cells (P = 0.006), gamma delta T cells (P = 0.007), monocytes (P = 0.047), and resting myeloid dendritic cells (P = 0.031). The low ELFN1-AS1 expression group had higher numbers of CD8+ T cells (P = 0.001) and neutrophils (P = 0.024) (Fig. 13b). Correlation analysis was performed to determine the co-expression patterns among 22 tumor-infiltrating immune cell types (Supplementary Figure S4).

Violin plots for tumor-infiltrating immune cells. a Comparison of immune cell proportions between high and low MIR4435-2HG expression groups. b Comparison of immune cell proportions between high and low ELFN1-AS1 expression groups. The blue bars represent the low expression groups, and the red bars represent the high expression groups
Discussion
Colorectal cancer (CRC) is one of the leading causes of cancer-related death worldwide. Increasing evidence has shown that dysregulated lncRNAs or lncRNA-mediated ceRNA networks are involved in the progression and prognosis of the CRC. LncRNAs suppress miRNA-induced translational inhibition of gene expression by competitively binding to miRNA response elements. In recent years, they have gained wide attention due to their important roles in tumor progression, patient prognosis, and immune infiltration [7, 13, 23]. The lncRNA H19/miR-29b-3p/PGRN axis promotes EMT in CRC and counts a great deal for seeking appropriate diagnostic biomarkers and therapeutic targets for CRC [11]. The oncogenic HOTAIR/miR-331-3p/HER2 axis is associated with tumor progression and poor prognosis in gastric cancer [24]. In addition, the RP11-1094 M14.8/miR-1269a/CXCL9 axis is a potential prognostic marker for gastric cancer with different degrees of immune cell infiltration [13]. Thus, identifying lncRNAs and their associated ceRNA networks as prognostic factors may provide a foundation for the development of early diagnostic biomarkers and therapeutic targets in CRC.
In this study, we analyzed TCGA sequence data from CRC patients and constructed a prognostic ceRNA network to explore the potential function of lncRNAs and their related genes. MIR4435-2HG and ELFN1-AS1 were identified as potential prognostic biomarkers and therapeutic targets in CRC based on survival and clinical feature analysis. MIR4435-2HG promotes CRC growth and metastasis through the miR-206/YAP1 axis and predicts poor prognosis in CRC patients [25,26,27]. ELFN1-AS1 is elevated in CRC tissues, and its upregulation promotes CRC cell proliferation and migration and inhibits apoptosis by competing with the endogenous miR-4644/TRIM44 axis [28, 29]. While downregulation of ELFN1-AS1 suppresses the activation of extracellular signal-regulated protein kinase (ERK), decreases vimentin expression, and increases E-cadherin expression.
Based on these ceRNA networks and lncRNA-mRNA co-expression relationships, we obtained two novel ceRNA regulatory subnetworks for MIR4435-2HG and ELFN1-AS1. According to the univariate and multivariate Cox analysis, ELFN1-AS1 was identified as an independent prognostic factor for CRC patients. MIR4435-2HG positively regulated PLAU, VCAN, LOX, COL5A2, and OSBPL3 mRNA expression by acting as a miRNA sponge. PLAU upregulation in primary tumor tissues is positively correlated with distant metastasis and indicates poor patient prognosis in CRC [30]. MiR-193a inhibits CRC cell growth and progression partly by downregulating the expression of target gene PLAU, which is consistent with our prediction of PLAU/miR-193a interaction [31]. VCAN is overexpressed in various human malignancies, including CRC [32,33,34,35,36,37], and CRC patients with high VCAN expression have worse disease outcomes than those with lower expression [38]. Upregulation of LOX expression is correlated with the progression and metastasis of various solid tumors [39]. There have been contradictory reports regarding the role of LOX in CRC. Csiszar et al. [40] reported that reduced LOX expression in colon tumor tissue supports the function of LOX as a tumor suppressor gene. However, recent studies found that LOX expression was significantly increased in tumor tissue and correlated with poor clinical outcomes of CRC patients [41, 42]. COL5A2 is upregulated in CRC tissue compared to normal tissue, suggesting that COL5A2 is involved in CRC carcinogenesis [43]; however, there are no reports about the role of OSBPL3 in CRC. E2F1 and SRM were predicted to be co-expressed genes involved in the ELFN1-AS1 regulatory network. Several studies have demonstrated that E2F1 is an oncogene involved in CRC proliferation and metastasis. Higher E2F1 expression levels indicate a shorter OS in CRC patients [44, 45]. However, there is also evidence to support E2F1 as a tumor suppressor gene involved in the induction of apoptosis in CRC [46, 47]. SRM encodes one of the key enzymes in the polyamine biosynthesis pathway. Polyamines are essential for cell proliferation and growth, and high expression of polyamines has been linked to several tumors, including CRC [48,48,49,50,51,52]. However, the direct correlation between the expression of SRM and human tumorigenesis remains unclear.
Subsequently, we performed GSEA analysis to explore the enriched biological pathways of the co-expressed genes. Most were enriched in the “extracellular matrix component,” “angiogenesis,” and “cell adhesion molecule binding,” which correlated with cancer pathogenesis. The extracellular matrix (ECM) is the main component of the tumor microenvironment and is composed of proteins, glycoproteins, proteoglycans, and polysaccharides [53]. Disorders in the ECM structure and composition promote cellular transformation and metastasis during cancer progression [54]. Angiogenesis is the formation of new blood vessels from existing vasculature, an important event in tumor growth, invasion into surrounding tissue, and distant metastasis [55]. Inhibition of angiogenesis has been a valuable therapeutic strategy for many solid tumors. Cell adhesion molecules (CAMs) play an important role in the interaction of cells with other cells and their surrounding intercellular environment. The role of CAMs in cancer involves angiogenesis, disruption of tissue architecture, and loss of contact with other cells and the ECM [56, 57].
Tumor-infiltrating immune cells (TIICs) include T cells, dendritic cells (DCs), B cells, macrophages, neutrophils, mast cells, and natural killer (NK) cells. They are major components of the complex microenvironment that regulates tumor progression and predicts prognosis in CRC [58,59,60]. To further explore the immune infiltration-related mechanism of how ceRNA regulates CRC prognosis, the estimated proportions of 22 tumor-infiltrating immune cell types in CRC were evaluated by CIBERSORT analysis based on MIR4435-2HG and ELFN1-AS1 expression levels. Our results revealed that nine immune cell types, including CD4+ memory resting T cells, CD4+ memory activated T cells, CD8+ T cells, gamma delta T cells, follicular helper T cells, plasma B cells, neutrophils, monocytes, and resting myeloid DCs, showed statistical differences between the high and low expression groups. Several recent studies reported that the type, density, and location of tumor-infiltrating immune cells are associated with favorable outcomes in CRC [58, 61]. The densities of CD4+ and CD8 + memory T cells were negatively correlated with local tumor invasion based on the T stage of the TNM classification [59]. DCs are potent antigen-presenting cells that play an important role in initiating, programming, and regulating antitumor immune responses. DC-based immunotherapy has been used as anti-cancer immunotherapy to treat various cancers, including CRC [62, 63]. The peripheral neutrophil-to-lymphocyte ratio (NLR) and lymphocyte-to-monocyte ratio (LMR) are prognostic factors for CRC patients [64, 65]. The association of MIR4435-2HG and ELFN1-AS1 lncRNA expression with tumor-infiltrating immune cell types supports their role as valuable prognostic markers and immunotherapeutic targets in CRC.
Nevertheless, there are still some limitations to the present study. First, the data used in this study were mainly from Western countries and lacked data from the Asian population. Second, the number of normal cases was relatively small, and a larger number of paired samples are needed for further analysis. Third, the multiple sources of the online verification data need to be further confirmed by homogeneous research samples and systematic experimental studies. For example, although the immunohistochemistry data were extracted from The Human Protein Atlas (THPA) to verify the upregulation of the identified genes in CRC at the protein level, the exact molecular mechanisms involving these hub genes have not been experimentally investigated in the present study. In addition, we observed opposing PLAU expression patterns at the mRNA and protein levels (Fig. 10 and Fig. 11), which is inconsistent with previous reports [66, 67]. Taken together, further experimental studies based on a larger sample size are needed to verify our hypotheses.
Conclusions
The regulatory networks of MIR4435-2HG and ELFN1-AS1 are considered to be closely associated with CRC prognosis. Although further functional experiments are required, our findings contribute to the development of MIR4435-2HG and ELFN1-AS1 as potential prognostic biomarkers and therapeutic targets in CRC.
Availability of data and materials
The data information of this study was obtained from the Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov/), the Human Protein Atlas (THPA; https://www.proteinatlas.org) and the GTEx projects.
Abbreviations
- lncRNA:
-
Long noncoding RNA
- miRNA:
-
MicroRNA
- mRNA:
-
Message RNA
- ceRNA:
-
Competing endogenous RNA
- TCGA:
-
The Cancer Genome Atlas
- DElncRNA:
-
Differentially expressed lncRNA
- DEmiRNA:
-
Differentially expressed miRNA
- DEmRNA:
-
Differentially expressed mRNA
- GEPIA:
-
Gene Expression Profiling Interactive Analysis
- GSEA:
-
Gene Set Enrichment Analysis
- CRC:
-
Colorectal cancer
- OS:
-
Overall survival
- EMT:
-
Epithelial-to-mesenchymal transition
- PPI:
-
Protein–protein interaction
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- STRING:
-
Search Tool for the Retrieval of Interacting Genes
- GTEx:
-
Genotype-Tissue Expression
- THPA:
-
The Human Protein Atlas
- NES:
-
Normalized enrichment score
- COAD:
-
Colon adenocarcinoma
- READ:
-
Rectal adenocarcinoma
- ERK:
-
Extracellular-signal regulated protein kinase
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Dekker E, Tanis PJ, Vleugels J, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019;394(10207):1467–80.
Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43(6):904–14.
Chai J, Guo D, Ma W, Han D, Dong W, Guo H, Zhang Y. A feedback loop consisting of RUNX2/LncRNA-PVT1/miR-455 is involved in the progression of colorectal cancer. Am J Cancer Res. 2018;8(3):538–50.
Wang XD, Lu J, Lin YS, Gao C, Qi F. Functional role of long non-coding RNA CASC19/miR-140-5p/CEMIP axis in colorectal cancer progression in vitro. World J Gastroenterol. 2019;25(14):1697–714.
Cui M, Chen M, Shen Z, Wang R, Fang X, Song B. LncRNA-UCA1 modulates progression of colon cancer through regulating the miR-28-5p/HOXB3 axis. J Cell Biochem. 2019.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? CELL. 2011;146(3):353–8.
Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. CELL. 2018;172(3):393–407.
Wang Y, Lu Z, Wang N, Feng J, Zhang J, Luan L, Zhao W, Zeng X. Long noncoding RNA DANCR promotes colorectal cancer proliferation and metastasis via miR-577 sponging. Exp Mol Med. 2018;50(5):1–17.
Pei J, Xiao W, Zhu D, Ji X, Shi L, Deng X. LncRNA DSCAM-AS1 promotes proliferation, migration and invasion of colorectal Cancer cells via modulating miR-144-5p/CDKL1. Life Sci. 2019;117050.
Ding D, Li C, Zhao T, Li D, Yang L, Zhang B. LncRNA H19/miR-29b-3p/PGRN Axis promoted epithelial-Mesenchymal transition of colorectal Cancer cells by acting on Wnt signaling. Mol Cells. 2018;41(5):423–35.
Chen DL, Lu YX, Zhang JX, Wei XL, Wang F, Zeng ZL, Pan ZZ, Yuan YF, Wang FH, Pelicano H, et al. Long non-coding RNA UICLM promotes colorectal cancer liver metastasis by acting as a ceRNA for microRNA-215 to regulate ZEB2 expression. THERANOSTICS. 2017;7(19):4836–49.
Zhang K, Zhang L, Mi Y, Tang Y, Ren F, Liu B, Zhang Y, Zheng P. A ceRNA network and a potential regulatory axis in gastric cancer with different degrees of immune cell infiltration. Cancer Sci. 2020;111(11):4041–50.
Karagkouni D, Paraskevopoulou MD, Tastsoglou S, Skoufos G, Karavangeli A, Pierros V, Zacharopoulou E, Hatzigeorgiou AG. DIANA-LncBase v3: indexing experimentally supported miRNA targets on non-coding transcripts. Nucleic Acids Res. 2020;48(D1):D101–10.
Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Database issue):D146–52.
Hsu SD, Lin FM, Wu WY, Liang C, Huang WC, Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39(Database issue):D163–9.
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–98.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102.
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419.
Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, Liu XS. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509–14.
Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. NATURE. 2014;505(7483):344–52.
Liu XH, Sun M, Nie FQ, Ge YB, Zhang EB, Yin DD, Kong R, Xia R, Lu KH, Li JH, et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer. 2014;13:92.
Dong X, Yang Z, Yang H, Li D, Qiu X. Long non-coding RNA MIR4435-2HG promotes colorectal Cancer proliferation and metastasis through miR-206/YAP1 Axis. Front Oncol. 2020;10:160.
Shen MY, Zhou GR, Y ZZ. LncRNA MIR4435-2HG contributes into colorectal cancer development and predicts poor prognosis. Eur Rev Med Pharmacol Sci. 2020;24(4):1771–7.
Ouyang W, Ren L, Liu G, Chi X, Wei H. LncRNA MIR4435-2HG predicts poor prognosis in patients with colorectal cancer. Peer J. 2019;7:e6683.
Lei R, Feng L, Hong D. ELFN1-AS1 accelerates the proliferation and migration of colorectal cancer via regulation of miR-4644/TRIM44 axis. Cancer Biomark. 2019.
Dong L, Ding C, Zheng T, Pu Y, Liu J, Zhang W, Xue F, Kang P, Ma Y, Wang X, et al. Extracellular vesicles from human umbilical cord mesenchymal stem cells treated with siRNA against ELFN1-AS1 suppress colon adenocarcinoma proliferation and migration. Am J Transl Res. 2019;11(11):6989–99.
Yang JL, Seetoo D, Wang Y, Ranson M, Berney CR, Ham JM, Russell PJ, Crowe PJ. Urokinase-type plasminogen activator and its receptor in colorectal cancer: independent prognostic factors of metastasis and cancer-specific survival and potential therapeutic targets. Int J Cancer. 2000;89(5):431–9.
Lin M, Zhang Z, Gao M, Yu H, Sheng H, Huang J. MicroRNA-193a-3p suppresses the colorectal cancer cell proliferation and progression through downregulating the PLAU expression. Cancer Manag Res. 2019;11:5353–63.
Miquel-Serra L, Serra M, Hernandez D, Domenzain C, Docampo MJ, Rabanal RM, de Torres I, Wight TN, Fabra A, Bassols A. V3 versican isoform expression has a dual role in human melanoma tumor growth and metastasis. Lab Investig. 2006;86(9):889–901.
Ricciardelli C, Brooks JH, Suwiwat S, Sakko AJ, Mayne K, Raymond WA, Seshadri R, LeBaron RG, Horsfall DJ. Regulation of stromal versican expression by breast cancer cells and importance to relapse-free survival in patients with node-negative primary breast cancer. Clin Cancer Res. 2002;8(4):1054–60.
Du WW, Yang BB, Shatseva TA, Yang BL, Deng Z, Shan SW, Lee DY, Seth A, Yee AJ. Versican G3 promotes mouse mammary tumor cell growth, migration, and metastasis by influencing EGF receptor signaling. PLoS One. 2010;5(11):e13828.
Ricciardelli C, Russell DL, Ween MP, Mayne K, Suwiwat S, Byers S, Marshall VR, Tilley WD, Horsfall DJ. Formation of hyaluronan- and versican-rich pericellular matrix by prostate cancer cells promotes cell motility. J Biol Chem. 2007;282(14):10814–25.
Casey RC, Oegema TJ, Skubitz KM, Pambuccian SE, Grindle SM, Skubitz AP. Cell membrane glycosylation mediates the adhesion, migration, and invasion of ovarian carcinoma cells. Clin Exp Metastasis. 2003;20(2):143–52.
Yoon H, Liyanarachchi S, Wright FA, Davuluri R, Lockman JC, de la Chapelle A, Pellegata NS. Gene expression profiling of isogenic cells with different TP53 gene dosage reveals numerous genes that are affected by TP53 dosage and identifies CSPG2 as a direct target of p53. Proc Natl Acad Sci U S A. 2002;99(24):15632–7.
Liu X, Xu D, Liu Z, Li Y, Zhang C, Gong Y, Jiang Y, Xing B. THBS1 facilitates colorectal liver metastasis through enhancing epithelial-mesenchymal transition. Clin Transl Oncol. 2020.
Nishioka T, Eustace A, West C. Lysyl oxidase: from basic science to future cancer treatment. Cell Struct Funct. 2012;37(1):75–80.
Csiszar K, Fong SF, Ujfalusi A, Krawetz SA, Salvati EP, Mackenzie JW, Boyd CD. Somatic mutations of the lysyl oxidase gene on chromosome 5q23.1 in colorectal tumors. Int J Cancer. 2002;97(5):636–42.
Wei B, Zhou X, Liang C, Zheng X, Lei P, Fang J, Han X, Wang L, Qi C, Wei H. Human colorectal cancer progression correlates with LOX-induced ECM stiffening. Int J Biol Sci. 2017;13(11):1450–7.
Baker AM, Cox TR, Bird D, Lang G, Murray GI, Sun XF, Southall SM, Wilson JR, Erler JT. The role of lysyl oxidase in SRC-dependent proliferation and metastasis of colorectal cancer. J Natl Cancer Inst. 2011;103(5):407–24.
Fischer H, Stenling R, Rubio C, Lindblom A. Colorectal carcinogenesis is associated with stromal expression of COL11A1 and COL5A2. CARCINOGENESIS. 2001;22(6):875–8.
Qian W, Zhang Z, Peng W, Li J, Gu Q, Ji D, Wang Q, Zhang Y, Ji B, Wang S, et al. CDCA3 mediates p21-dependent proliferation by regulating E2F1 expression in colorectal cancer. Int J Oncol. 2018;53(5):2021–33.
Suzuki T, Yasui W, Yokozaki H, Naka K, Ishikawa T, Tahara E. Expression of the E2F family in human gastrointestinal carcinomas. Int J Cancer. 1999;81(4):535–8.
Dosil MA, Navaridas R, Mirantes C, Tarragona J, Eritja N, Felip I, Urdanibia I, Megino C, Domingo M, Santacana M, et al. Tumor suppressive function of E2F-1 on PTEN-induced serrated colorectal carcinogenesis. J Pathol. 2019;247(1):72–85.
Zacharatos P, Kotsinas A, Evangelou K, Karakaidos P, Vassiliou LV, Rezaei N, Kyroudi A, Kittas C, Patsouris E, Papavassiliou AG, et al. Distinct expression patterns of the transcription factor E2F-1 in relation to tumour growth parameters in common human carcinomas. J Pathol. 2004;203(3):744–53.
Upp JJ, Saydjari R, Townsend CJ, Singh P, Barranco SC, Thompson JC. Polyamine levels and gastrin receptors in colon cancers. Ann Surg. 1988;207(6):662–9.
Luk GD, Goodwin G, Marton LJ, Baylin SB. Polyamines are necessary for the survival of human small-cell lung carcinoma in culture. Proc Natl Acad Sci U S A. 1981;78(4):2355–8.
Manni A, Grove R, Kunselman S, Aldaz CM. Involvement of the polyamine pathway in breast cancer progression. Cancer Lett. 1995;92(1):49–57.
Gupta S, Ahmad N, Marengo SR, MacLennan GT, Greenberg NM, Mukhtar H. Chemoprevention of prostate carcinogenesis by alpha-difluoromethylornithine in TRAMP mice. Cancer Res. 2000;60(18):5125–33.
Gilmour SK. Polyamines and nonmelanoma skin cancer. Toxicol Appl Pharmacol. 2007;224(3):249–56.
Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395–406.
Crotti S, Piccoli M, Rizzolio F, Giordano A, Nitti D, Agostini M. Extracellular matrix and colorectal Cancer: how surrounding microenvironment affects Cancer cell behavior? J Cell Physiol. 2017;232(5):967–75.
Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29(6 Suppl 16):15–8.
Lewczuk Ł, Pryczynicz A, Guzińska-Ustymowicz K. Cell adhesion molecules in endometrial cancer - a systematic review. Adv Med Sci. 2019;64(2):423–9.
Lafrenie RM, Buckner CA, Bewick MA. Cell adhesion and cancer: is there a potential for therapeutic intervention? Expert Opin Ther Targets. 2007;11(6):727–31.
Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. ONCOGENE. 2008;27(45):5904–12.
Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306.
Ye L, Zhang T, Kang Z, Guo G, Sun Y, Lin K, Huang Q, Shi X, Ni Z, Ding N, et al. Tumor-infiltrating immune cells act as a marker for prognosis in colorectal Cancer. Front Immunol. 2019;10:2368.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–4.
Hassannia H, Ghasemi CM, Atyabi F, Nosouhian M, Masjedi A, Hojjat-Farsangi M, Namdar A, Azizi G, Mohammadi H, Ghalamfarsa G, et al. Blockage of immune checkpoint molecules increases T-cell priming potential of dendritic cell vaccine. Immunology. 2020;159(1):75–87.
Kajihara M, Takakura K, Kanai T, Ito Z, Saito K, Takami S, Shimodaira S, Okamoto M, Ohkusa T, Koido S. Dendritic cell-based cancer immunotherapy for colorectal cancer. World J Gastroenterol. 2016;22(17):4275–86.
Mizuno R, Kawada K, Itatani Y, Ogawa R, Kiyasu Y, Sakai Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int J Mol Sci. 2019;20:3.
Tan D, Fu Y, Tong W, Li F. Prognostic significance of lymphocyte to monocyte ratio in colorectal cancer: a meta-analysis. Int J Surg. 2018;55:128–38.
Kohga S, Harvey SR, Weaver RM, Markus G. Localization of plasminogen activators in human colon cancer by immunoperoxidase staining. Cancer Res. 1985;45(4):1787–96.
Harvey SR, Sait SN, Xu Y, Bailey JL, Penetrante RM, Markus G. Demonstration of urokinase expression in cancer cells of colon adenocarcinomas by immunohistochemistry and in situ hybridization. Am J Pathol. 1999;155(4):1115–20.
Acknowledgements
We thank the editors from BioMed Proofreading LLC for comprehensive editing of this manuscript (Additional file 5).
Funding
This research was supported by the National Natural Science Foundation of China (Nos. 30872923 and 81672853) and Peking University People’s Hospital Scientific Research Development Funds (RDH2020–11). The funding bodies had no role in the design of the study, the collection, analysis, or interpretation of data, or the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
JYC, YXS, and WDY designed the study. ML, YZ, and TRL downloaded and analyzed data. JYC, JS, and DW wrote the initial version of the manuscript. YLL, JZG and WDY revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there is no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Clustered heatmaps of differentially expressed RNAs. (A) Heatmaps of lncRNAs. (B) Heatmaps of miRNAs. (C) Heatmaps of mRNAs.
Additional file 2.
Construction of a survival-related ceRNA network. Red nodes represent the upregulated RNAs, and blue nodes represent the downregulated RNAs.
Additional file 3.
Univariate and multivariable Cox regression analysis of RNAs involved in the ceRNA network.
Additional file 4.
Correlation of 22 tumor-infiltrating immune cell types.
Additional file 5.
Certificate of English copyediting service.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, J., Song, Y., Li, M. et al. Comprehensive analysis of ceRNA networks reveals prognostic lncRNAs related to immune infiltration in colorectal cancer. BMC Cancer 21, 255 (2021). https://doi.org/10.1186/s12885-021-07995-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-07995-2