
Abstract
Background
Crystal-storing histiocytosis (CSH) is a rare lesion characterized by sheets of crystal-laden non-neoplastic histiocytes. CSH shows a prominent association with lymphoproliferative disorders that express monoclonal immunoglobulins, mainly multiple myeloma (MM), lymphoplasmacytic lymphoma (LPL) and monoclonal gammopathy of undetermined significance (MGUS). However, no aggressive B cell lymphoma has been reported to be associated with CSH.
Case presentation
A 74-year-old Chinese woman presented with multiple subcutaneous masses, abdominal pain, and fever. An IgM kappa type of monoclonal gammopathy (MG) was noted by immunofixation performed on the patient’s serum. Computed tomographic (CT) scan revealed subcutaneous masses on the left upper arm and at the waist and multiple low-density lesions in the spleen. Microscopically, sections of subcutaneous masses revealed sheets of large polygonal and spindle cells with abundant eosinophilic cytoplasm, round to ovoid eccentric nuclei, reticulate chromatin, and median nucleoli. Massive needle-shaped crystals were confined to the cytoplasm. Immunohistochemically, these crystal-containing cells were positive for CD68/PGM1, CD163, IgM, and Igκ. Meanwhile, the splenic tumour was diagnosed as diffuse large B-cell lymphoma (DLBCL), non-germinal-centre B (non-GCB) subtype (Hans algorithm). Immunohistochemistry for IgM was positive in the cytoplasm of some neoplastic cells. Immunoglobulin heavy chain (IgH) gene rearrangement was detected by PCR analysis of the subcutaneous mass and the splenic tumour.
Conclusion
To the best of our knowledge, this is the first case of generalized CSH with DLBCL and MG. Although the rarity of CSH and separate locations of CSH and lymphoma led to a diagnostic dilemma, the presence of MG was a clue to appreciate the relation between CSH and DLBCL. This case stressed a full investigation into the underlying lymphoproliferative disorder for integrated diagnosis and correct treatments.
Similar content being viewed by others


Background
Crystal-storing histiocytosis (CSH) is a rare lesion characterized by sheets of crystal-laden non-neoplastic histiocytes. Characteristic crystalline inclusions within the cytoplasm of histiocytes were first described by Glaus in 1917 [1]. The condition is still under-recognized given the rare incidence. Notably, CSH shows a prominent association with an underlying lymphoproliferative disorder, mainly MM, LPL and MGUS [2]. However, no aggressive B cell lymphoma has been reported to be associated with CSH. We report a case of CSH with DLBCL in a 74-year-old Chinese woman who presented with subcutaneous masses, abdominal pain, and fever. IgM κ paraproteinemia and proteinuria were also present. To the best of our knowledge, this is the first report of CSH with DLBCL and MG.
Case presentation
Medical history
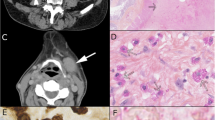
A 74-year-old woman was admitted with the chief complaint of subcutaneous masses, abdominal pain, and fever. Six months before the admission, the patient developed thrombocytopenia while being treated with antibiotics for pneumonia. At the time of admission, physical examination revealed a firm non-tender subcutaneous mass on the left upper arm (Fig. 1a) and two at the waist. Her abdomen was soft, but light tenderness was present in the upper abdomen without rebound tenderness. The liver and the spleen were not palpable below the costal margin. The laboratory tests results were as follows. (1) A white blood cell count of 3.48 × 109/L (reference range 3.5–9.5 × 109/L), an extremely low platelet count of 7.0 × 109/L (reference range 100–300 × 109/L) and haemoglobin of 96 g/L (reference range 115–150 g/L) were noted. (2) Serum protein electrophoresis showed a monoclonal band, which was determined to be of the IgM κ type by immunofixation (Fig. 2a). Serum protein quantitative analysis revealed 10.30 g/L IgM, 20.10 g/L kappa light chain, 7.04 g/L lambda light chain and kappa/lambda = 2.86 (reference range 2.20,13.00 and 6.50 g/L and reference ratio of 2.56, respectively). All the other immunoglobin (Ig) levels were normal. (3) Urinalysis result was 1 + (0.5 g/L) (reference range negative, 0 g/L) for protein. (4) Bone marrow aspiration and flow cytometry (FCM) analysis were normocellular. Computed tomographic (CT) scan showed a 2.0 cmx1.6 cm subcutaneous mass on the left upper arm and several ill-defined soft tissue lesions at the waist along with intra-abdominal lymphadenopathy and moderate splenomegaly (Fig. 1b&c). Abdominopelvic contrast-enhanced CT displayed multiple low-density plaques in the spleen (Fig. 1d). The patient underwent a needle biopsy of the subcutaneous mass on the left arm in a local hospital. After her admission, the subcutaneous mass at the right waist was excised, and laparoscopic splenectomy was performed.

General observation and imaging examinations. (a): Subcutaneous mass at the left upper arm. (b-d): CT scans before the treatment. (b): A 2.0 cmx1.6 cm subcutaneous poor-defined tumour (arrow) at the left upper arm. (c): Several ill-defined soft tissue density lesions (arrows) at the waist. (d): Multiple low-density plaques (arrow) in the enlarged spleen by abdominopelvic contrast-enhanced CT. (e&f): Smaller lumbar masses (arrows) by PET-CT at the nine-month follow-up

Serum protein studies. (a): Before the treatment, serum protein electrophoresis showed a monoclonal band, which was determined to be of the IgM κ type by immunofixation. (b): The paraprotein disappeared at the one-year follow-up
Pathologic findings
(1) Macroscopically, the resected lumbodorsal specimen was a firm grey-brown mass measuring 5.5 cm × 3.0 cm × 1.5 cm in volume. Microscopic examination revealed diffuse proliferation in the subcutaneous tissue, which was composed of large polygonal and spindle histiocytes with abundant eosinophilic cytoplasm, round to ovoid eccentric nuclei, reticulate chromatin and median nucleoli (Figs. 3a&b). Needle-shaped crystals were confined to the cytoplasm, some of which were in parallel arrays (Fig. 3c). These crystal-containing cells were located in and around an intact lymph node measuring 0.8 cm in maximal dimension. The paracortical area of the lymph node was expanded by extensive infiltration of the crystal-containing cells and mature plasma cells (Fig. 3a). These crystal-containing cells were strongly positive for CD68/PGM1 (Fig. 3d) and CD163 and negative for S-100, desmin, SMA, MSA, and myogenin. Immunostain for IgM heavy chain was strong and diffuse (Fig. 3e). Immunostain for the κ light chain was relatively weak. Immunostains for the IgG heavy chain and λ light chain were negative (Fig. 3f). The plasma cells were characterized by CD38+, MUM1+, and kappa > lambda. Congo red staining was negative. BIOMED-2 multiplex PCR analysis showed diversity-joining gene segments of immunoglobin heavy chain (IgH-DH-JH) gene rearrangement (the monoclonal peak was at approximately 400 bp) and Igκ gene rearrangement. The MYD88 L265p mutation was not detected. The diagnosis of “CSH associated with IgH and Igκ rearrangements” was made with a comment that an underlying lymphoproliferative disorder should be suspected.

Microscopic appearance and immunophenotypes of the subcutaneous masses at the waist. (a): The lesion is restricted in the subcutaneous tissue and an intact lymph node infiltrated by crystal-laden histiocytes (the pink area), HE × 10. (b): Sheets of large polygonal and spindle histiocytes, HE × 400. (c): Needle-shaped crystals within the cells, HE × 1000. (d): CD68 × 400; (e): IgM × 1000; (f): IgG × 400
(2) Review of the needle biopsy of the mass on the left upper arm identified a similar cell population as the one at the waist, which was characterized by sheets of large rhabdoid cells with abundant deeply eosinophilic cytoplasm containing massive crystalline materials, eccentric irregular nuclei, and inconspicuous nucleoli.
(3) The spleen was enlarged to 15 cm × 10 cm × 8 cm and weighed 198 g with a smooth surface. Serial sectioning showed a grey irregular solid tumour measuring 3 cm × 1.5 cm × 1.3 cm at the splenic hilum mixed with adipose tissue and focal necrotic areas. Microscopic examination revealed diffuse infiltration of large cells with a round, oval or irregular nuclei, distinct nucleoli and scant cytoplasm, centroblast like (Fig. 4a-b). Frequent mitoses were observed (Fig. 4b). Immunohistochemically, the neoplastic cells were CD20+, Bcl6+, MUM1+, CD3p-, CD10-, CyclinD1- and CD43- (Fig. 4c-d). IgM was strongly positive in the cytoplasm of some neoplastic cells, while IgG, CD38, and CD138 were negative (Fig. 4e-g). The Ki-67 index was 80% (Fig. 4h). Fluorescence in situ hybridization (FISH) suggested BCL-6 rearrangements but the absence of dysregulations of BCL2 and MYC. In situ hybridization for Epstein-Barr virus-encoded RNA (EBER-ISH) was negative. A monoclonal peak of the IgH gene was detected at approximately 420 bp by commercial BIOMED-2 multiplex PCR system. These findings conformed to a diagnosis of DLBCL, non-GCB subtype (Hans algorithm). The pancreas was involved, while the liver was not. CSH lesions were not found in the spleen and the regional lymph nodes.

Microscopic appearance and immunophenotypes of the tumour at the splenic hilum. (a-h): Formalin-fixed paraffin-embedded splenic tumour sections (a): A focal infiltration of lymphocytes, HE × 10. (b): Diffuse large centroblast-like cells with a round, oval or irregular nuclei, distinct nucleoli, and scant cytoplasm. Frequent mitoses were seen. HE × 400. (c-h) original magnification, × 400. (c): CD20; (d): MUM-1; (e): IgM (Insert × 1000); (f): IgG; (g): CD138; (h): Ki-67
Treatment and follow-up
After splenectomy, the platelet count increased to 128 × 109/L. The patient was administered eight courses of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) and was in partial remission (PR) 9 months after diagnosis. Positron emission tomography-computed tomography (PET-CT) scan showed resolution of the mass at the left arm, smaller lumbar subcutaneous masses and intraperitoneal lymphadenopathy (Fig. 1e-f). However, serum paraprotein levels remained elevated (IgM 4.01 g/L). Thus, the patient received four cycles of rituximab (600 mg/m2 day 1) plus lenalidomide (25 mg/m2 days 1–14 q28), and the MG was not visible (Fig. 2b) at a one-year follow-up.
Discussion and conclusions
CSH is a rare disorder diagnosed only on the basis of pathologic features, but its prominent association with lymphoproliferative disorders has been expounded in the literature. To date, 140 cases of CSH have been reported according to a PubMed search of the English literature. Patients can be classified into localized CSH (L-CSH) and generalized CSH (G-CSH) based on the number of the foci. G-CSH, which entails the involvement of two or more organs/systems, may present with a rapid clinical course and portend a worse prognosis [2]. Recently, Fang H et al. reported 8 cases of CSH with bone marrow involvement and reviewed the English literature (total of 131 cases) from 1987 to 2017 [3]. Of the 131 cases of CSH, 100 cases (76%) were associated with a lymphoproliferative disorder, mainly MM, LPL, and MGUS. Thirty-five cases (27%) were accompanied with B-cell lymphoma, but none of these lymphomas were aggressive B-cell lymphoma. To the best of our knowledge, this is the first case of CSH with DLBCL.
DLBCL was mentioned in 5 patients with CSH in the literature (Table 1), and four of these cases transformed from or were accompanied with low-grade B-cell lymphoma [4,5,6,7]. Although a concurrent low-grade B-cell component cannot be completely excluded in the unbiopsied sites of abdominal lymphadenopathy in the present case, biopsies of multiple anatomic sites failed to obtain any evidence of low-grade B-cell lymphoma, and normal bone marrow biopsy and the undetected MYD88 mutation contributed to the exclusion of LPL. In addition, there was no history of lymphoproliferative disorders in our patient. Kawano et al. reported a case of pulmonary L-CSH without lymphoproliferative disorders in a patient with a history of gastric DLBCL [8]. In that report, the gastric DLBCL retained CR without evidence of recurrence, and the PCR analysis with stomach and lung tissues did not reveal the same clone. Thus, the ultimate diagnosis was L-CSH without lymphoproliferative disorders. Similarly, in the present case, the CSH lesions and the DLBCL involved different anatomic sites respectively and exhibited unrelated IgH gene monoclonal peaks in PCR analysis. However, the presence of MG and the generalized CSH raised concerns for a systematic association. As expected, subsequent immunohistochemistry showed strong IgM staining in some DLBCL cells without plasmablastic differentiation. These neoplasm cells may be responsible for the IgM type of MG and the generalized CSH. In addition, the level of serum paraprotein changed with the response to chemotherapy, and this finding could point to the unusual origin of immunoglobulins. Taking these findings together, we preferred a diagnosis of CSH with DLBCL rather than a CSH independent of the DLBCL and MG.
The mechanism of crystal formation is unclear. It is hypothesized that a combination of overproduction and conformational alterations in the immunoglobin light chains leads to crystallization, impaired enzymatic degradation by histiocytes, and crystal accumulation within histiocytes [9,10,11]. Afterwards, Kanagal-Shamanna et al. observed that the sole representation of the variable region of immunoglobin heavy chains contributed to abnormal immunoglobin structure by proteomic methods, expanding the proposed hypothesis [12]. The present case posed a diagnostic challenge due to the separate locations of the CSH and DLBCL, and gene-rearrangement studies showed that the variable region encoded by IgH-DH-JH gene segments may be related to the crystallization. More molecular analyses are required to clarify the mechanism.
It is morphologically difficult to distinct CSH from other histiocytosis and sedimentary lesions, such as xanthogranuloma, Langerhans cell histiocytosis, granular cell tumour, fibrous histiocytoma, amyloidosis, and Gaucher’s disease. On low-power images, CSH is characterized by sheets of polygonal or spindle-shaped histiocytes with abundant eosinophilic cytoplasm. On high-power images, refractive needle-like crystalline material filled the cytoplasm. Immunohistochemical analysis is useful to the differential diagnosis. In our case, CD68 and CD163 immunohistochemical stains confirmed that the large, pink cells were histiocytes. S100 protein immunohistochemical stain excluded the possibility of granular cell tumour and Langerhans cell histiocytosis. Congo red excluded amyloidosis, and desmin, MSA and myogenin were performed to exclude adult rhabdomyoma. There were a few cases of CSH mimicking adult rhabdomyoma given the similar morphologic features and location in soft tissue [13,14,15]. In patients with a malignant tumour, CSH may be clinically diagnosed as metastatic carcinoma [16].
Finally, the treatment and prognosis of patients with CSH vary depending on the associated disease [2]. Regarding DLBCL, MG was reported as a poor prognostic marker in patients with the non-GCB type [17]. The outcome after R-CHOP was unsatisfactory in IgM-secreting DLBCL patients, while immunochemotherapy combined with bortezomib or lenalidomide seemed to be more effective [18]. In our patient, the general status improved with R-CHOP. However, the serum paraprotein remained elevated, and multiple lymph node involvement remained detectable. Lenalidomide accelerated the disappearance of paraprotein and might play a positive role in this subset of patients. Further accumulation of cases is essential to expound the treatment and prognosis of CSH and DLBCL with MG.
In conclusion, the presented case is the first to describe G-CSH concomitant with DLBCL and MG. The rarity of CSH and separate locations of CSH and DLBCL contributed to the complexity of the diagnostic approach. This case highlights an unusual origin of immunoglobulins and the overproduction of the immunoglobulins may promote the G-CSH. Immunohistochemical analysis can assist in differential diagnosis and classification. Once a pathologic diagnosis of CSH is confirmed, subsequent investigations for the underlying lymphoproliferative disorders are required, such as a detailed clinical history, serum and urine protein studies, imaging examinations and bone marrow biopsy.
Availability of data and materials
All the data supporting the findings are presented within the manuscript and supplementary data.
Abbreviations
- BM:
-
Bone marrow
- CR:
-
Complete remission
- CSH:
-
Crystal-storing histiocytosis
- CT:
-
Computed tomographic
- DH-JH:
-
diversity-joining gene segments of the immunoglobin heavy chain
- DLBCL:
-
Diffuse large B-cell lymphoma
- EBER-ISH:
-
In situ hybridization for Epstein-Barr virus-encoded RNA
- F:
-
Female
- FCM:
-
Flow cytometry
- FISH:
-
Fluorescence in situ hybridization
- G-CSH:
-
Generalized crystal-storing histiocytosis
- Ig:
-
Immunoglobulin
- IgH :
-
Immunoglobulin heavy chain
- L-CSH:
-
Localized crystal-storing histiocytosis
- LN:
-
Lymph node
- LPL:
-
Lymphoplasmacytic lymphoma
- LP-PCD:
-
Lymphoproliferative or plasma cell disorder
- M:
-
Male
- MG:
-
Monoclonal gammopathy
- MGUS:
-
Monoclonal gammopathy of undetermined significance
- MM:
-
Multiple myeloma
- mo:
-
Month; y:Year
- non-GCB:
-
non-germinal-center B
- PCR:
-
Polymerase chain reaction
- PET-CT:
-
Positron emission tomography–computed tomography
- PR:
-
Partial remission
- WM:
-
Waldenström’s macroglobulinemia
References
Glaus A. On multiple myelocytoma with peculiar crystalline cell structures, combined with elastolysis and extensive amyloidosis and calcification. Virchows Arch A. 1917;223(3):301–39.
Dogan S, Barnes L, Cruz-Vetrano WP. Crystal-storing histiocytosis: report of a case, review of the literature (80 cases) and a proposed classification. Head and neck pathology. 2012;6(1):111–20.
Fang H, Chiu A, Reichard KK. Crystal-storing Histiocytosis in bone marrow: a Clinicopathologic study of eight cases and review of the literature. Am J Clin Pathol. 2018;149(2):148–63.
Jones D, Bhatia VK, Krausz T, Pinkus GS. Crystal-storing histiocytosis: a disorder occurring in plasmacytic tumors expressing immunoglobulin kappa light chain. Hum Pathol. 1999;30(12):1441–8.
Kaufmann O, Hansen A, Deicke P, Burmester GR, Dietel M. Subcutaneous crystal-storing histiocytosis associated with lymphoplasmacytic lymphoma (immunocytoma). Pathol Res Pract. 1996;192(11):1148–51.
Li JJ, Henderson C. Cutaneous crystal storing histiocytosis: a report of two cases. J Cutan Pathol. 2015;42:136–43.
Thorson P, Hess JL. Transformation of monocytoid B-cell lymphoma to large cell lymphoma associated with crystal-storing histiocytes. Arch Pathol Lab Med. 2000;124(3):460–2.
Kawano N, Beppu K, Oyama M, Himeji D, Yoshida S, Kuriyama T, Ono N, Masuyama H, Yamashita K, Yamaguchi K, et al. Successful surgical treatment for pulmonary crystal-storing histiocytosis following the onset of gastric non-hodgkin lymphoma. J Clin Exp Hematop. 2013;53(3):241–5.
Lebeau A, Zeindl-Eberhart E, Muller EC, Muller-Hocker J, Jungblut PR, Emmerich B, Lohrs U. Generalized crystal-storing histiocytosis associated with monoclonal gammopathy: molecular analysis of a disorder with rapid clinical course and review of the literature. Blood. 2002;100(5):1817–27.
Aline-Fardin A, Bender S, Fabiani B, Buob D, Brahimi S, Verpont MC, Mothy M, Ronco P, Boffa JJ, Aucouturier P, et al. Pseudo-peritoneal Carcinomatosis presentation of a crystal-storing Histiocytosis with an Unmutated monoclonal kappa light chain. Medicine. 2015;94(32):e1247.
El Hamel C, Thierry A, Trouillas P, Bridoux F, Carrion C, Quellard N, Goujon JM, Aldigier JC, Gombert JM, Cogne M, et al. Crystal-storing histiocytosis with renal Fanconi syndrome: pathological and molecular characteristics compared with classical myeloma-associated Fanconi syndrome. Nephrology, dialysis, transplantation: official publication of the European Dialysis and transplant association. European Renal Association. 2010;25(9):2982–90.
Kanagal-Shamanna R, Xu-Monette ZY, Miranda RN, Dogan A, Zou D, Luthra R, Weber DM, O'Malley DP, Jorgensen JL, Khoury JD, et al. Crystal-storing histiocytosis: a clinicopathological study of 13 cases. Histopathology. 2016;68(4):482–91.
Li ZS, Li PF, Wang Z, Huang GS. Primary extranodal soft-tissue B-cell lymphoma with abundant immunoglobulin inclusions mimicking adult rhabdomyoma: a case report. J Med Case Rep. 2011;5:53.
Friedman MT, Molho L, Valderrama E, Kahn LB. Crystal-storing histiocytosis associated with a lymphoplasmacytic neoplasm mimicking adult rhabdomyoma: a case report and review of the literature. Arch Pathol Lab Med. 1996;120(12):1133–6.
Kapadia SB, Enzinger FM, Heffner DK, Hyams VJ, Frizzera G. Crystal-storing histiocytosis associated with lymphoplasmacytic neoplasms. Report of three cases mimicking adult rhabdomyoma. Am J Surg Pathol. 1993;17(5):461–7.
Balakrishna J, Chen A, Urken M. Crystal storing histiocytosis clinically mimicking metastatic carcinoma: report of a case and reviews of literature. Head & neck. 2016;38(4):E95–8.
Kim YR, Kim SJ, Cheong JW, Kim Y, Jang JE, Lee JY, Min YH, Song JW, Yang WI, Kim JS. Monoclonal and polyclonal gammopathy measured by serum free light chain and immunofixation subdivide the clinical outcomes of diffuse large B-cell lymphoma according to molecular classification. Ann Hematol. 2014;93(11):1867–77.
Cox MC, et al. Clinicopathologic characterization of diffuse-large-B-cell lymphoma with an associated serum monoclonal IgM component. PLoS One. 2014;9(4):e93903.
Acknowledgments
We appreciate Dr. Hou Li to treat this patient.
Funding
This study was supported by a grant of Key Program founded by Health and Family Planning Commission of Sichuan Province(NO.16ZD009)and a grant of Application Basic Research Project founded by Science & Technology Department of Sichuan Province (NO.2017JY0266). Funders provided the financial support for sample testing but did not participate in the design and implementation of the project and the writing and submission of articles.
Author information
Authors and Affiliations
Contributions
LWP made substantial contributions to supply the case and to design the research work. YJQ and GLM made substantial contributions to the molecular testing and interpretation of data. GLM, CZH, WM, and XCX made substantial contributions to the acquisition of clinical data and their interpretation. TQ wrote the initial draft of the manuscript. LWP, ZWY, and CZH have been involved in revising the paper for important intellectual content. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
As it is a case report, ethics approval is not necessary after consulting the Ethics Committee of West China Hospital.
Consent for publication
Written consent has been obtained from the patient for her clinical details and images to be published in this study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tao, Q., Zhang, W., Chen, Z. et al. Generalized crystal-storing histiocytosis with diffuse large B-cell lymphoma and monoclonal gammopathy in a Chinese elderly woman: a case report. BMC Cancer 19, 514 (2019). https://doi.org/10.1186/s12885-019-5734-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-019-5734-x