
Abstract
Background
Shikonin, a natural naphthoquinone, is abundant in Chinese herb medicine Zicao (purple gromwell) and has a wide range of biological activities, especially for cancer. Shikonin and its analogues have been reported to induce cell-cycle arrest, but target information is still unclear. We hypothesized that shikonin, with a structure similar to that of quinone-type compounds, which are inhibitors of cell division cycle 25 (Cdc25) phosphatases, will have similar effects on Cdc25s. To test this hypothesis, the effects of shikonin on Cdc25s and cell-cycle progression were determined in this paper.
Methods
The in vitro effects of shikonin and its analogues on Cdc25s were detected by fluorometric assay kit. The binding mode between shikonin and Cdc25B was modelled by molecular docking. The dephosphorylating level of cyclin-dependent kinase 1 (CDK1), a natural substrate of Cdc25B, was tested by Western blotting. The effect of shikonin on cell cycle progression was investigated by flow cytometry analysis. We also tested the anti-proliferation activity of shikonin on cancer cell lines by MTT assay. Moreover, in vivo anti-proliferation activity was tested in a mouse xenograft tumour model.
Results
Shikonin and its analogues inhibited recombinant human Cdc25 A, B, and C phosphatase with IC50 values ranging from 2.14 ± 0.21 to 13.45 ± 1.45 μM irreversibly. The molecular modelling results showed that shikonin bound to the inhibitor binding pocket of Cdc25B with a favourable binding mode through hydrophobic interactions and hydrogen bonds. In addition, an accumulation of the tyrosine 15-phosphorylated form of CDK1 was induced by shikonin in a concentration-dependent manner in vitro and in vivo. We also confirmed that shikonin showed an anti-proliferation effect on three cancer cell lines with IC50 values ranging from 6.15 ± 0.46 to 9.56 ± 1.03 μM. Furthermore, shikonin showed a promising anti-proliferation effect on a K562 mouse xenograph tumour model.
Conclusion
In this study, we provide evidence for how shikonin induces cell cycle arrest and functions as a Cdc25s inhibitor. It shows an anti-proliferation effect both in vitro and in vivo by mediating Cdc25s.
Similar content being viewed by others

Background
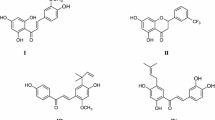
Dual-specificity protein phosphatases (DSP) Cdc25s (Cdc25A, Cdc25B, and Cdc25C) have an essential role in cell cycle progression via controlling the phosphorylation state of their natural substrates cyclin-dependent kinases (CDKs). Overexpression of Cdc25s and over-activation of CDKs are involved in tumour-associated cell-cycle aberration [1]. Therefore, Cdc25s is currently considered to be a promising anticancer target [2,3,4]. Several inhibitors of Cdc25s with antitumour activity have been reported [5, 6]. Most potent small molecule inhibitors of the Cdc25 phosphatases are quinone-derived compounds, such as menadione (vitamin K3) [7], compound 2 [8], NSC95397 [9], compound 5 [10], and NSC668394 [11] (Fig. 1). This type of compound was hypothesized to inhibit the activity of Cdc25s by oxidation of the catalytically essential cysteine residue in the enzyme’s active site through production of reactive oxygen species [12].

Structures of quinone-type inhibitors of Cdc25s and shikonin
The naphthoquinone-type natural product shikonin was derived from the root of Lithospermum erythrorhizon, which has broad applications in Traditional Chinese Medicine [13, 14]. Over the past few decades, a number of studies have demonstrated multiple biological effects of shikonin. It has been reported to have anti-HIV [14], anti-inflammatory [15, 16], antibacterial, and anticancer [17,18,19,20] activities. Among these activities, the anticancer activity, especially the induction of apoptosis and necroptosis, is well reported [13, 19,20,21,22,23]. However, the key target is still unclear. Shikonin has a similar chemical skeleton to that of the quinone-type inhibitors of Cdc25s. Therefore, we hypothesized that shikonin will have similar effects on Cdc25s. To test this hypothesis, the effects of shikonin on Cdc25s and related biofunction were confirmed in this paper.
Methods
Chemicals
Shikonin and its analogues are natural products that were purchased from Herbest, Inc. (Baoji, Shanxi, China). All other chemicals were purchased from Sigma-Aldrich (Shanghai, China) unless otherwise noted.
Measurement of phosphatase inhibitory activity of shikonin and its analogues
CycLex® protein phosphatase Cdc25A, -B and -C fluorometric assay Kit (CycLex, Cat. No. CY-1352, CY-1353, CY-1354) was used to test the enzyme inhibition rate of shikonin and its analogues for Cdc25A, -B and -C. In summary, dual-specificity phosphatase activity was measured in a 96-well microtiter plate using O-methylfluorescein phosphate (OMFP) as a substrate. 5 μL (0.1 μg/μL) of purified recombinant Cdc25s (Cdc25A, -B and -C) was mixed with 40 μL of assay mixture and incubated with 5 μL of the test compound at various concentrations in a well. Then, 25 μL of stop solution was added. Fluorescence was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using a fluorescence microplate reader (BioTek Instruments, Inc., Winooski, VT, USA). Menadione is a quinone-type inhibitor of Cdc25s [7] that was used as a positive control here.
For the dialysis assay, the enzyme-inhibitor complex including 0.2 μM Cdc25B and 50 μM shikonin was dialyzed against 5000-fold of the assay buffer for the indicated period of time. At the end of each dialysis, Cdc25B activity was determined as described above.
Molecular modelling
The docking method used is described in a previous work [24]. In summary, molecular modelling was performed using Maestro 9.0. The X-ray structure of Cdc25B (PDB ID: 1QB0) was downloaded from the Protein Data Bank (PDB, https://www.rcsb.org) and prepared with the “Protein Preparation Wizard” workflow with default settings. The grid-enclosing box was generated within 10 Å of Cys473 in the refined crystal structure. The ligand structure was prepared with the Ligprep module. Finally, shikonin was docked into Cdc25B using Glide (version 5.5) in extra precision (XP) mode with default settings [25]. Favourable binding poses were selected according to the docking score and view check.
Cell lines and culture conditions
K562 cells (myelogenous leukaemia cell line), MCF-7 cells (breast cancer cell line) and HeLa cells (cervical cancer cells) were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China). The catalogue numbers of these cell lines are TCHu191, TCHu74 and TCHu187, respectively. The temperature-sensitive FT210 cell line (tsFT210) is a mouse breast cancer cell line that is widely used for studying cell cycle progression. Intracellular CDK1 protein of tsFT210 is inactive at 39 °C because of two point mutations on the cdc2 gene, which leads to it being easily controlled at different cell cycle phases through changes in temperature [26]. This cell line was kindly provided by Dr. Rongcai Yue from his laboratory (School of Pharmacy, Second Military Medical University). All cell lines were kept in the logarithmic growth phase in 5% CO2 at 37 °C with RPMI-1640 medium supplemented with 10% FBS and 1% penicillin G-streptomycin in a humidified chamber at 5% CO2.
Western blotting
The phosphorylation status of CDK1 was analysed by Western blotting as described in our previous work [24]. In summary, tsFT210 cells (1 × 106) were treated with shikonin (0, 1, 5 and 25 μM) for 4 h followed by collection and suspension in lysis buffer (150 mM NaCl, 50 mM Tris, 0.02% NaN3, 0.01% phenylmethylsulfonyl fluoride, 0.2% aprotinin, and 1% TritonX-100, pH 8.0) containing a protease inhibitor cocktail (Thermo Scientific) for 30 min at 4 °C. The protein concentration in cell lysates was confirmed by the Bradford method. Fifty micrograms of protein per lane was resolved on 10% SDS polyacrylamide gels, and the bands were transferred to PVDF membranes (Millipore). Nonspecific reactivity was blocked by 5% non-fat milk prepared in TBST (10 mM Tris, 150 mM NaCl, 0.05% Tween-20, pH 7.5) at room temperature for 1 h. The protein signals were captured with primary antibodies and the secondary antibodies according to the manufacturers’ instructions. In this process, the protein β-actin was used to normalize target protein. All antibodies used in this paper were purchased from Cell Signaling Technology (Inc, China).
Intracellular reactive oxygen species assay
Reactive oxygen species (ROS) were detected with the fluorescent probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA) as described by others [18, 27]. Briefly, MCF-7 cells or tsFT210 (synchronized at G2 phase) cells were plated in 96-well plates and loaded with 20 μM DCFH-DA for 30 min at 37 °C in culture medium. Cells were washed with PBS three times after incubation and then treated with shikonin at concentrations of 5 μM and 25 μM for 4 h. Untreated cells were used as control. Fluorescence was measured using a fluorescence microplate reader (BioTek Instruments, excitation wavelength: 485 nm, emission wavelength: 528 nm). The ROS levels were expressed as RFU (relative fluorescence unit).
Cell cycle analysis
Cell cycle analysis was performed as described previously [28]. Briefly, tsFT210 cells were plated at 1 × 105 cells/well (24-well plate) and maintained at 32 °C. Cell proliferation was blocked at the G2 phase by increasing the temperature from 32 to 39 °C and treating for 17 h. Then, the cells were released at 32 °C and immediately treated with shikonin. The harvested cells were stained with staining buffer (50 μg/ml propidium iodide, 0.1% sodium citrate and 0.2% NP-40) and analysed by Flow Cytometry (BD Biosciences). A final concentration of 0.5% DMSO was used for all compounds and as a negative control. Nocodazole, a potent mitotic blocker that arrests cells at the G2/M phase, was used as a positive control at a concentration of 100 nM.
Anti-proliferation assay
The anti-proliferation activity of shikonin against cancer cells was tested by the MTT method in 96-well plates. First, 4 × 103 cells were seeded per well and treated with the shikonin with serial concentrations for 48 h. Then, the cells were incubated with 10 μL of MTT solution (5 mg/mL in PBS) for an additional 2–4 h at 37 °C. The formazan crystals were dissolved with 100 μL of DMSO after removing the supernatant. The absorbance at 570 nm was measured by a BioTek Synergy 2 plate reader (BioTek Instruments, Inc., Winooski, VT, USA). Doxorubicin, a chemotherapy medication used to treat cancer and having a broad spectrum of anticancer activities [29], was used as a positive control here.
Xenograph tumour model in mice
The mouse model was constructed according to our previous work [24]. Balb/c mice (female, 5 weeks old), purchased from Shanghai SLAC Laboratory Animal Co., LTD (Shanghai, China), were subcutaneously injected into a 100 μL K562 cell suspension (5 × 106/mL, PBS) in the right flank of mice. We started treating the mice with shikonin at 1, 5, or 10 mg/kg or vehicle containing 5% DMSO (control group) by intraperitoneal injection every other day when the tumour’s volume reached approximately 200 mm3. The long and short dimensions of tumour size were measured by a digital calliper at days 1, 4, 8, 11, 16, 21, and 24 after treatment. The volume was calculated with the formula (long dimension) × (short dimension)2/2. The mice were euthanized 2 days after the last treatment by the method of cervical dislocation.
Statistical analysis
Statistical analyses were performed using PRISM software. All quantitative values are given as the means ± SEM. An unpaired Student’s t test was used to evaluate the difference between two different treatments, and p < 0.05 was considered statistically significant. The statistical significance of differences in the survival of mice from the different groups was determined by the log-rank test using the same program.
Results
Shikonin induce irreversible inhibition of human recombinant CDC25 phosphatases
Using the protein phosphatase Cdc25 combo fluorometric assay kit, shikonin and its analogues in vitro inhibited recombinant human Cdc25A, -B, and -C phosphatases in a concentration-dependent manner with IC50 values ranging from 2.14 to 14.32 μM (Table 1). The positive control menadione led to an inhibition of 4.12 ± 0.87, 5.37 ± 0.45, and 5.13 ± 0.24 μM, respectively.
Previous research revealed that the quinone-type inhibitors of Cdc25 phosphatases play a role through irreversible oxidation of the catalytic cysteine of Cdc25B [30, 31]. To identify the inhibition pattern of shikonin, dialysis was used to study the reversibility of shikonin action. As shown in Fig. 2a, after dialysis for 2, 4, 6, and even 24 h, the activity of Cdc25B was not rescued from inhibition by shikonin, indicating that the Cdc25B was inhibited irreversibly. In addition, to confirm whether the shikonin has similar mechanism with other quinone-type inhibitors, the ROS content was measured in cells treated with or without shikonin. After treating with shikonin, intracellular ROS in MCF-7 and tsFT210 cells increased significantly compared with those of control cells (Fig. 2b), suggesting that Cdc25s inhibition by shikonin could be caused by redox cycling of the shikonin similar to that of other quinones.

Shikonin inhibits Cdc25B irreversibly in vitro and induces ROS production in MCF-7 cells. a Activity of shikonin-treated Cdc25B after dialysis. b ROS production in MCF-7 and tsFT210 cells after treatment with shikonin. Data points represent fold change of three independent experiments. *P < 0.05; **P < 0.01 compared with the control group
Molecular model of shikonin interactions with the Cdc25B catalytic domain
In order to evaluate the binding mode and affinity of shikonin with Cdc25B, molecular modelling was performed using the docking program Glide. X-ray crystallography showed that the catalytic domain of Cdc25B contains the canonical HCX5R PTPase catalytic-site motif [32]. The phosphorylated amino acids of the substrate usually bind to this loop through forming hydrogen bonds to arginine R. The quinone-type inhibitor was supposed to bind the inhibitor binding pocket, which was beside the active site (Fig. 3), and induce oxidation of the catalytic cysteine through redox cycling reaction [1]. After docking the shikonin into the above binding loop around cys473, we found that the shikonin bound to the inhibitor binding pocket similar to other quinone-type inhibitors. Figure 2 shows the most energetically favourable binding pose from docking results. In such a binding mode, the benzene ring of shikonin is involved in hydrophobic interactions with the pocket residues. In addition, a hydrogen bond between one oxygen of the naphthoquinone core and Tyr428 was predicted.

Binding mode of shikonin in the Cdc25B binding cavity. Shikonin (yellow) and key interacting residues (green) are represented as stick models with the protein as a surface. H-bonds are shown as dashed black lines. The figure was generated using Pymol from Protein Data Bank ID 1QB0
Shikonin inhibits cell cycle progression
Inhibition of Cdc25s results in hyper-phosphorylation of CDKs and cell cycle arrest. Therefore, the impact of shikonin on cell cycle progress was investigated. The tsFT210 cell line has been widely used for studying cell cycle progression because it can be easily controlled at different cell cycle phases through changing temperature [26]. The synchronized tsFT210 cells were then treated with the indicated concentration of shikonin and nocodazole (a potent mitotic blocker) or 1% DMSO for 6 h. The positive control nocodazole arrested the cells at the G2/M phase. Comparatively, the cells treated with shikonin were blocked at the G2/M phase in a concentration-dependent manner, whereas the DMSO-treated control cells had completed mitosis and had entered the following cell cycle (Fig. 4). Such results indicate that shikonin can target and delay cell cycle progression at the G2/M phase.

Cell cycle analysis of tsFT210 cells in the absence or presence of shikonin. a G2/M-arrested cells after a temperature shift for 17 h at 39 °C. b DMSO-treated cells after a temperature shift for 4 h at 32 °C. c Cells treated with 100 nM nocodazole. d-f Cells treated with 1–25 μM shikonin. Data are representative of two independent experiments
Shikonin inhibits CDK1 dephosphorylation
Endogenous Cdc25s controls the cell cycle through dephosphorylation of their natural substrate CDKs [2]. Therefore, CDK1 protein will be hyper-phosphorylated if the CDC25s are inhibited. In order to confirm whether shikonin inhibits the activity of intracellular Cdc25 phosphatases, the phosphorylation status of CDK1 was analysed by Western blotting. As shown in Fig. 5, shikonin at 1 μM, 5 μΜ, and 25 μM induced an accumulation of the tyrosine 15-phosphorylated form of CDK1. These results suggested that shikonin downregulated the activity of Cdc25s, leading to hyper-phosphorylation of CDK1 in cultured cells.

Inhibition of CDK1 dephosphorylation caused by shikonin. Cells in G2/M phase were treated with the indicated concentrations of shikonin and DMSO for 4 h and then harvested. Samples were processed for Western blot analysis. Data are representative of two independent experiments
Shikonin inhibits in vitro tumour cell proliferation
To evaluate the anti-proliferation activity of shikonin for different cancer cells, four cancer cell lines (MCF-7, HeLa, K562, and tsFT210) were selected to investigate the anti-proliferation activity. The viability of cells was evaluated by the MTT method. As shown in Table 2, shikonin markedly inhibits the proliferation of MCF-7 cells (IC50: 6.82 ± 0.76 μM), HeLa cells (IC50: 9.56 ± 1.03 μM), K562 cells (IC50: 6.15 ± 0.46 μM) and tsFT210 cells (8.97 ± 0.87 μM) compared with the negative control. Doxorubicin was used as a positive control and showed normal anticancer activity according to a previous report.
Shikonin has anti-tumour effects in vivo by inhibiting CDK1 dephosphorylation
Xenograph tumour model mice were used to evaluate if shikonin can inhibit tumour growth in vivo. K562-bearing mice were treated with shikonin at different doses and vehicle for 21 days. The tumour sizes were recorded in this process. As shown in Fig. 5a, shikonin concentration-dependently inhibited tumour growth. Higher doses of shikonin had a better inhibitory effect and longer observed survival time (Fig. 6a, 6b). In the process of this experiment, the tumour growth of mice from higher dose groups was significantly inhibited compared with those of the control and low-dose groups. These results suggest that shikonin has profound anticancer activity in vivo. Moreover, the protein levels of phosphorylated CDK1 in primary tumour tissues obtained from the mice were significantly increased after treatment by shikonin compared with that of the control group, especially for the 5 and 10 mg/kg groups (Fig. 6c). This means that the shikonin still participates in the cell cycle process in vivo.

Shikonin inhibits tumour growth in vivo in the K562-bearing mice by affecting the phosphorylation of CDK1 (n = 10/group). a Tumour volume plot of K562-bearing mice treated with vehicle or shikonin at 1, 5, or 10 mg/kg by oral gavage for 21 days. The tumours were measured twice per week. The data are represented as the mean ± SEM. Tumour growth was inhibited significantly after treatment with shikonin compared with the control group. *P < 0.05; †P < 0.01; ‡P < 0.001 compared with the control group. b Kaplan-Meier survival plot of the K562-bearing nude mice. Survival of the K562-bearing nude mice was prolonged in the shikonin-treated groups compared with control group. c The phosphorylation level of CDK1 is affected by shikonin.
Discussion
Overexpression of CDC25s, which leads to an irregular cell cycle, is often present in cancer cells. They control the cell cycle by activating CDKs. Therefore, CDC25s are considered attractive targets for anticancer drug discovery. Several potent CDC25s inhibitors have been identified; however, few of them have entered clinical trials. Shikonin, a natural naphthoquinone, has a similar structure to that of the well-reported quinone-type inhibitors of CDC25s. Hence, we hypothesized that shikonin will have similar effects on Cdc25s. To test this hypothesis, the effects of shikonin on Cdc25s and related biofunction were proved in this work.
Shikonin and six analogues showed considerable inhibition for CDC25s using OMFP as substrate. The main difference between these analogues and shikonin is the R groups (Table 1), but they have similar inhibition for CDC25s. Therefore, we suggest that the R groups have little effect on the activity and that the pharmacological group is the skeleton of naphthoquinone. Finally, shikonin was selected for further research because it showed the best inhibition for CDC25s compared with other analogues and can be generated from other analogues by hydrolysing the R group. Shikonin binds to the same pocket of Cdc25B as other quinone-type inhibitors according to the molecular modelling results. Together with the unaltered activity of Cdc25B after dialysis and increasing ROS content after treatment with shikonin, we suggested that shikonin inhibited Cdc25B similar to other quinone-type inhibitors, which were hypothesized to disrupt Cdc25s phosphatase activity by oxidation of the catalytically essential cysteine residue in the enzyme’s active site through production of reactive oxygen species [23].
Endogenous Cdc25s control the cell cycle through dephosphorylating their natural substrate CDKs. Therefore, the impacts of shikonin on cell cycle progress and phosphorylation state of CDK1 were investigated. We proved that shikonin could block the cell cycle of tsFT210 at the G2/M phase and effectively inhibit dephosphorylation of CDK1 in vitro.
We also evaluated the anti-proliferation activity of shikonin both in vitro and in vivo. Shikonin showed broad spectrum anticancer activity after evaluating the anti-proliferation activity for four cancer cell lines. Moreover, tumour growth of xenograph tumour model mice was significantly inhibited by shikonin. To confirm the in vivo activity of shikonin was through inhibition of the Cdc25s, we also checked the phosphorylation state of CDK1 in tumour tissue from xenograph tumour model mice, and the results showed that shikonin could inhibit dephosphorylation of CDK1 in tumour tissue also, which suggested that shikonin could also participate in the Cdc25s-related cell cycle pathway in vivo.
Shikonin reportedly exerts anticancer effects with less drug resistance [33], which can be attribute to its multiple targets. ROS generation, inactivation of NF-κB, upregulation of p53 and p21, and activation of caspases may be involved in the anticancer mechanisms of shikonin [34,35,36,37,38]. In brief, shikonin disrupts the cancer cell lines by inhibiting either the necroptosis or apoptosis pathway. Cancer cells died predominantly through the necroptotic pathway when they were co-treated with shikonin and the apoptosis inhibitor Z-VAD-FMK, while apoptosis became a selective route for cell death when the cancer cells were co-treated with shikonin and necroptotic inhibitor Nec-1 [39]. Moreover, shikonin and its analogues have been reported can induce cell cycle arrest, but the mechanism is still unclear [27]. Here, we identified shikonin as an inhibitor of Cdc25s that plays an essential role in cell cycle progression.
The normal development of tissue depends on the homeostasis and balance between cell proliferation and death (Fig. 7). When out of balance, as generated by inappropriate proliferation or inhibition of apoptosis and necroptosis pathways, cancer usually results [40]. If one pathway was inhibited or key proteins of it mutated, shikonin can also play an anticancer role through two other pathways, which could explain the lower drug resistance of shikonin.

Schematic of the coordination between cell proliferation and death. ↓: inhibited by shikonin, ↑: promoted by shikonin
Conclusions
In conclusion, shikonin was first identified as an inhibitor of Cdc25s in the current study. It was shown to inhibit cancer cell growth by arresting the cell cycle through binding to Cdc25s in vitro and in vivo. Together with previous research, shikonin is shown to have anticancer function through regulation of the apoptosis, necroptosis, and cell cycle pathways.
Change history
19 May 2023
A Correction to this paper has been published: https://doi.org/10.1186/s12885-023-10976-2
Abbreviations
- CDK:
-
Cyclin-dependent kinases
- ROS:
-
Reactive oxygen species
References
Kristjansdottir K, Rudolph J. Cdc25 phosphatases and cancer. Chemistry & biology. 2004;11(8):1043–51.
Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nature reviews Cancer. 2007;7(7):495–507.
Xing X, Chen J, Chen M. Expression of CDC25 phosphatases in human gastric cancer. Digestive diseases and sciences. 2008;53(4):949–53.
Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Current opinion in cell biology. 2006;18(2):185–91.
Lavecchia A, Di Giovanni C, Novellino E. CDC25 phosphatase inhibitors: an update. Mini reviews in medicinal chemistry. 2012;12(1):62–73.
Lavecchia A, Di Giovanni C, Novellino E. Inhibitors of Cdc25 phosphatases as anticancer agents: a patent review. Expert opinion on therapeutic patents. 2010;20(3):405–25.
Lee MH, Cho Y, Kim DH, Woo HJ, Yang JY, Kwon HJ, Yeon MJ, Park M, Kim SH, Moon C, et al. Menadione induces G2/M arrest in gastric cancer cells by down-regulation of CDC25C and proteasome mediated degradation of CDK1 and cyclin B1. Am J Transl Res. 2016;8(12):5246–55.
Brun MP, Braud E, Angotti D, Mondesert O, Quaranta M, Montes M, Miteva M, Gresh N, Ducommun B, Garbay C. Design, synthesis, and biological evaluation of novel naphthoquinone derivatives with CDC25 phosphatase inhibitory activity. Bioorg Med Chem. 2005;13(16):4871–9.
Lazo JS, Nemoto K, Pestell KE, Cooley K, Southwick EC, Mitchell DA, Furey W, Gussio R, Zaharevitz DW, Joo B, et al. Identification of a potent and selective pharmacophore for Cdc25 dual specificity phosphatase inhibitors. Molecular pharmacology. 2002;61(4):720–8.
Tamura K, Southwick EC, Kerns J, Rosi K, Carr BI, Wilcox C, Lazo JS. Cdc25 inhibition and cell cycle arrest by a synthetic thioalkyl vitamin K analogue. Cancer research. 2000;60(5):1317–25.
Lazo JS, Aslan DC, Southwick EC, Cooley KA, Ducruet AP, Joo B, Vogt A, Wipf P. Discovery and biological evaluation of a new family of potent inhibitors of the dual specificity protein phosphatase Cdc25. J Med Chem. 2001;44(24):4042–9.
Brisson M, Foster C, Wipf P, Joo B, Tomko RJ Jr, Nguyen T, Lazo JS. Independent mechanistic inhibition of cdc25 phosphatases by a natural product caulibugulone. Mol Pharmacol. 2007;71(1):184–92.
Gong K, Li W. Shikonin, a Chinese plant-derived naphthoquinone, induces apoptosis in hepatocellular carcinoma cells through reactive oxygen species: A potential new treatment for hepatocellular carcinoma. Free radical biology & medicine. 2011;51(12):2259–71.
Chen X, Yang L, Zhang N, Turpin JA, Buckheit RW, Osterling C, Oppenheim JJ, Howard OM. Shikonin, a component of chinese herbal medicine, inhibits chemokine receptor function and suppresses human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2003;47(9):2810–6.
Sun WX, Liu Y, Zhou W, Li HW, Yang J, Chen ZB. Shikonin inhibits TNF-alpha production through suppressing PKC-NF-kappaB-dependent decrease of IL-10 in rheumatoid arthritis-like cell model. J Nat Med. 2017;71(2):349–56.
Liao PL, Lin CH, Li CH, Tsai CH, Ho JD, Chiou GC, Kang JJ, Cheng YW. Anti-inflammatory properties of shikonin contribute to improved early-stage diabetic retinopathy. Scientific reports. 2017;7:44985.
Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011;30(42):4297–306.
Huang C, Luo Y, Zhao J, Yang F, Zhao H, Fan W, Ge P. Shikonin kills glioma cells through necroptosis mediated by RIP-1. PloS one. 2013;8(6):e66326.
Wada N, Kawano Y, Fujiwara S, Kikukawa Y, Okuno Y, Tasaki M, Ueda M, Ando Y, Yoshinaga K, Ri M, et al. Shikonin, dually functions as a proteasome inhibitor and a necroptosis inducer in multiple myeloma cells. Int J Oncol. 2015;46(3):963–72.
Kim HJ, Hwang KE, Park DS, Oh SH, Jun HY, Yoon KH, Jeong ET, Kim HR, Kim YS. Shikonin-induced necroptosis is enhanced by the inhibition of autophagy in non-small cell lung cancer cells. J Transl Med. 2017;15(1):123.
Zhang Z, Zhang Z, Li Q, Jiao H, Chong D, Sun X, Zhang P, Huo Q, Liu H. Shikonin induces necroptosis by reactive oxygen species activation in nasopharyngeal carcinoma cell line CNE-2Z. J Bioenerg Biomembr. 2017;49(3):265–72.
Shan ZL, Zhong L, Xiao CL, Gan LG, Xu T, Song H, Yang R, Li L, Liu BZ. Shikonin suppresses proliferation and induces apoptosis in human leukemia NB4 cells through modulation of MAPKs and cMyc. Mol Med Rep. 2017;16(3):3055–60.
Tang X, Zhang C, Wei J, Fang Y, Zhao R, Yu J. Apoptosis is induced by shikonin through the mitochondrial signaling pathway. Mol Med Rep. 2016;13(4):3668–74.
Zhang S, Yin J, Li X, Zhang J, Yue R, Diao Y, Li H, Wang H, Shan L, Zhang W. Jacarelhyperol A induced apoptosis in leukaemia cancer cell through inhibition the activity of Bcl-2 proteins. BMC cancer. 2014;14:689.
Garcia-Pineres AJ, Castro V, Mora G, Schmidt TJ, Strunck E, Pahl HL, Merfort I. Cysteine 38 in p65/NF-kappaB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J Biol Chem. 2001;276(43):39713–39,720.
Th’ng JP, Wright PS, Hamaguchi J, Lee MG, Norbury CJ, Nurse P, Bradbury EM. The FT210 cell line is a mouse G2 phase mutant with a temperature-sensitive CDC2 gene product. Cell. 1990;63(2):313–24.
Shahsavari Z, Karami-Tehrani F, Salami S, Ghasemzadeh M. RIP1K and RIP3K provoked by shikonin induce cell cycle arrest in the triple negative breast cancer cell line, MDA-MB-468: necroptosis as a desperate programmed suicide pathway. Tumour Biol. 2016;37(4):4479–91.
Tsuchiya A, Hirai G, Koyama Y, Oonuma K, Otani Y, Osada H, Sodeoka M. Dual-Specificity Phosphatase CDC25A/B Inhibitor Identified from a Focused Library with Nonelectrophilic Core Structure. ACS Med Chem Lett. 2012;3(4):294–8.
Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–70.
Brisson M, Nguyen T, Wipf P, Joo B, Day BW, Skoko JS, Schreiber EM, Foster C, Bansal P, Lazo JS. Redox regulation of Cdc25B by cell-active quinolinediones. Molecular pharmacology. 2005;68(6):1810–20.
Zhou YB, Feng X, Wang LN, Du JQ, Zhou YY, Yu HP, Zang Y, Li JY, Li J. LGH00031, a novel ortho-quinonoid inhibitor of cell division cycle 25B, inhibits human cancer cells via ROS generation. Acta Pharmacol Sin. 2009;30(9):1359–68.
Reynolds RA, Yem AW, Wolfe CL, Deibel MR Jr, Chidester CG, Watenpaugh KD. Crystal structure of the catalytic subunit of Cdc25B required for G2/M phase transition of the cell cycle. J Mol Biol. 1999;293(3):559–68.
Wu H, Xie J, Pan Q, Wang B, Hu D, Hu X. Anticancer agent shikonin is an incompetent inducer of cancer drug resistance. PloS one. 2013;8(1):e52706.
Wu Z, Wu L, Li L, Tashiro S-i, Onodera S, Ikejima T. p53-mediated cell cycle arrest and apoptosis induced by shikonin via a caspase-9-dependent mechanism in human malignant melanoma A375-S2 cells. J Pharmacol Sci. 2004;94(2):166–76.
Wu Z, Wu L-J, Li L-H, Tashiro S-I, Onodera S, Ikejima T. Shikonin regulates HeLa cell death via caspase-3 activation and blockage of DNA synthesis. J Asian Nat Prod Res. 2004;6(3):155–66.
Yeh C-C, Kuo H-M, Li T-M, Lin J-P, Yu F-S, Lu H-F, Chung J-G, Yang J-S. Shikonin-induced apoptosis involves caspase-3 activity in a human bladder cancer cell line (T24). in vivo. 2007;21(6):1011–9.
Min R, Zun Z, Min Y, Wenhu D, Wenjun Y, Chenping Z. Shikonin inhibits tumor invasion via down-regulation of NF-κB-mediated MMP-9 expression in human ACC-M cells. Oral diseases. 2011;17(4):362–9.
Chang I-C, Huang Y-J, Chiang T-I, Yeh C-W, Hsu L-S. Shikonin induces apoptosis through reactive oxygen species/extracellular signal-regulated kinase pathway in osteosarcoma cells. Biol Pharm Bull. 2010;33(5):816–24.
Shahsavari Z, Karami-Tehrani F, Salami S. Shikonin induced necroptosis via reactive oxygen species in the T-47D breast cancer cell line. Asian Pac J Cancer Prev. 2015;16:7261–6.
Hay MGBA. Cell proliferation and apoptosis. Current Opinion in Cell Biology. 1999;11:745.
Acknowledgements
Not applicable
Funding
This work was supported by the Project of Qinghai Science & Technology Department (2016-ZJ-Y01, 2018-ZJ-948Q) and the Open Project of State Key Laboratory of Plateau Ecology and Agriculture, Qinghai University (2017-ZZ-02). The first funding source was mainly used in the collection, analysis, and interpretation of experimental data. The second funding source mainly supported the drafting and revisions of the manuscript.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Author information
Authors and Affiliations
Contributions
Designed the experiments: SDZ, QG, SS and ZHS; Performed the experiments and analysed data: WL, QHS, SF, JMJ, LWZ and QQJ; Wrote the manuscript: SDZ and QG; Edited the manuscript: ZHS and SS; SDZ, QG, WL, QHS, SF, JMJ, LWZ, QQJ, SS, ZHS have given final approval of this version to be published.
Corresponding authors
Ethics declarations
Ethics approval
All cell lines used in this work are non-primary cells and do not required ethics approval for their use. All animal experiments were approved by the animal care committee of the Second Military Medical University in accordance with institutional and Chinese government guidelines for animal care and use.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, S., Gao, Q., Li, W. et al. Shikonin inhibits cancer cell cycling by targeting Cdc25s. BMC Cancer 19, 20 (2019). https://doi.org/10.1186/s12885-018-5220-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-018-5220-x