
Abstract
Background
Ataxia telangiectasia mutated (ATM) gene plays a key role in response to DNA lesions and is related to the invasion and metastasis of malignancy. Epidemiological studies have indicated associations between ATM rs1801516 polymorphism and different types of cancer, but their results are inconsistent. To further evaluate the effect of ATM rs1801516 polymorphism on cancer risk, we conducted this meta-analysis.
Methods
Studies were identified according to specific inclusion criteria by searching PubMed, Web of Science, and Embase databases. Pooled odds ratios (ORs) and corresponding 95% confidence intervals (CIs) under recessive, dominant, codominant, and overdominant models of inheritance were calculated to estimate the association between rs1801516 polymorphism and cancer risk.
Results
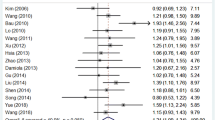
A total of 37 studies with 12,879 cases and 18,054 controls were included in our study. No significant association was found between rs1801516 polymorphism and cancer risk in overall comparisons (AA vs GG + GA: OR = 0.91, 95% CI, 0.78–1.07; AA+GA vs GG: OR = 1.00, 95% CI, 0.90–1.11; AA vs GG: OR = 0.89, 95% CI, 0.75–1.06; GA vs GG: OR = 1.01, 95% CI, 0.91–1.13; GG + AA vs GA: OR = 1.00, 95% CI, 0.88–1.10). However, after subgroup analyses by region-specified population, significant associations were found in European (AA vs GG + GA: OR = 0.79, 95% CI, 0.65–0.96, P = 0.017; AA vs GG: OR = 0.79, 95% CI, 0.65–0.96, P = 0.017), South American (AA+GA vs GG: OR = 2.15, 95% CI, 1.37–3.38, P = 0.001; GA vs GG: OR = 2.19, 95% CI, 1.38–3.47, P = 0.001; GG + AA vs GA: OR = 0.46, 95% CI, 0.29–0.72, P = 0.001), and Asian (AA vs GG + GA: OR = 7.45, 95% CI, 1.31–42.46, P = 0.024; AA vs GG: OR = 7.40, 95% CI, 1.30–42.19, P = 0.024). Subgroup analyses also revealed that compared with subjects carrying a GG genotype, those carrying a homozygote AA had a decreased risk for breast cancer (AA vs GG: OR = 0.76, 95% CI, 0.59–0.98, P = 0.035), and the homozygote AA was associated with decreased cancer risk in subjects with family history (AA vs GG: OR = 0.68, 95% CI, 0.47–0.98, P = 0.039).
Conclusions
ATM rs1801516 polymorphism is not associated with overall cancer risk in total population. However, for subgroup analyses, this polymorphism is especially associated with breast cancer risk; in addition, it is associated with overall cancer risk in Europeans, South Americans, Asians, and those with family history.
Similar content being viewed by others

Background
Cancer is a worldwide public health problem, and considerable parts of death are due to cancer every year. It is reported that one fourth deaths in the United States is caused by cancer [1]. According to the latest cancer data from the GLOBOCAN website, there were 14.1 million new cancer cases, 8.2 million cancer deaths, and 32.6 million people living with cancer (within 5 years of diagnosis) in 2012 worldwide [2]. The statistical data of cancer in 2017 shows that 1,688,780 new cancer cases (836,150 males and 852,630 females) are expected to be diagnosed in the United States, and 600,920 Americans (318,420 males and 282,500 females) are expected to die of cancer [3]. For all sites combined, both the incidence rate and death rate are higher in males than those in females, and the most commonly diagnosed cancers are lung cancer, prostate cancer, breast cancer, and colon cancer [3].
Pathogenesis of cancer has been studied worldwide for a long time, generating different theories, such as the gene mutation, oxidative stress, and ionization radiation theories. Single nucleotide polymorphisms (SNPs) on different genes have been detected for finding specific biomarkers in different cancers. Ataxia telangiectasia-mutated (ATM) gene is one of the most frequently studied genes in cancer occurrence and progression. Mutation on ATM leads to the human autosomal recessive disorder, ataxia-telangiectasia (A-T), resulting in high cellular radiosensitivity, chromosomal instability, immunodeficiency, and cancer predisposition [4, 5]. Lymphomas and leukemia are predominant in all types of cancer in A-T patients, and the cancer incidence rate in black A-T patients is as more than two times as that in whites [6, 7]. ATM gene is located in human chromosome 11q22–23, spans over 160 kb DNA, and encodes a 315 kDa protein. As a member belonging to the phosphoinositide 3-kinase (PI3-K)-related protein kinase family, ATM is activated by a series of cellular stress events, such as DNA double-strand break (DSB), reactive oxygen species, hypotonic stress, and chloroquine [8]. ATM is involved in important life processes, including DNA repair, cell cycle regulation, neuroprotection, immunity, metabolism, longevity, and fertility [8].
Several ATM polymorphism loci have been studied in different types of cancer, including rs1801516, which is a common nonsynonymous variant on this gene. Genome-wide association studies (GWAS) have identified rs1801516 as a susceptibility locus for melanoma [9]. Large-sample case-control studies have assessed effects of this polymorphism on risk of breast cancer, prostate cancer, rectal cancer, bladder cancer, lung cancer, pancreatic cancer, and thyroid cancer. Meta-analyses have also been performed to assess ATM rs1801516 polymorphism and cancer predisposition, but the results are inconsistent [10,11,12,13,14].
We performed this meta-analysis to further identify the association between rs1801516 polymorphism and cancer risk using larger sample size than ever before, and using the trial sequential analysis (TSA) to give more comprehensive conclusions.
Methods
We conducted this meta-analysis according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines [15].
Search strategy
Systematic search of publications was performed in PubMed, Web of Science, and Embase datasets (last search on November 18, 2017). Because of different nomenclatures for SNP, we took all the names that might be used in different studies of this SNP into consideration in our searching terms: “(rs1801516 or G5557A or 5557G>A or 5557 G/A or Asp1853Asn or D1853N or G1853A) and (cancer or carcinoma or malignancy)”.
Inclusion criteria
Studies included in this meta-analysis met the following criteria: (1) A human study with full text available; (2) A study on ATM rs1801516 polymorphism and cancer risk; (3) Using a case-control study design; (4) Using healthy subjects without malignant diseases as controls; (5) Genotype data is sufficient for odds ratio (OR) and 95% confidence interval (CI) estimation. In addition, we screened the reference lists of all the relevant studies, including eligible studies, reviews and meta-analyses, and only original articles published in English were included.
Data extraction
For each included study, the following information was extracted: the first author, year of publication, country, region-specified population, cancer type, source of controls, matching criteria of controls, family history, genotyping method, Hardy-Weinberg equilibrium (HWE) in controls, minor allele frequency (MAF) in cases and controls, sample size, and numbers of cases and controls with different genotype. Region-specified population in our meta-analysis was defined geographically as European, North American, South American, Asian, and Oceanian. Population-based controls (PBC) and hospital-based controls (HBC) were classified in our meta-analysis: blood donors and controls recruited from birth cohort, general population, and community are defined as PBC; and controls recruited from hospitals, clinics, research institutions, and biorepository were defined as HBC.
Quality assessment
Two authors (YG and JS) assessed the quality of each study independently according to the Newcastle-Ottawa Scale (NOS) for case-control studies [16]. A study can be awarded a maximum score of 9: 4 assigned for selection, 2 for comparability, and 3 for exposure. When inconsistency existed between the two authors, the third author (SQ) was requested to reassess the score of quality.
Statistical analysis
Allele and genotype frequencies in controls were calculated for each study to evaluate the HWE using chi-square test. Association between rs1801516 polymorphism and cancer risk was assessed by OR and corresponding 95% CI calculated from logistic regression. For each analysis, stratified or pooled, five comparisons were conducted, including dominant model (GA/AA vs GG), codominant model (GA vs GG and AA vs GG), recessive (AA vs GG/GA), and overdominant model (GA vs GG/AA). For studies of Sommer SS et al. [17], Gonzalez-Hormazabal P et al. [18], Maillard S et al. [19], and Calderon-Zuniga Fdel C et al. [20], no AA genotype was detected in either case or control group; thus, these studies were excluded in comparisons of AA vs GG and AA vs GA/GG. For studies of Yang H et al. [21], Bretsky P et al. [22], and Hirsch AE et al. [23], frequencies of GG and GA genotypes were presented together as GG/GA; thus, only association under recessive model was evaluated for these studies. For study of Xu L et al. [24], frequencies of GA and AA genotypes were presented together as GA/AA; thus, only association under dominant model was evaluated for this study. Subgroup analyses were performed by cancer type, region-specified population, source of control, matching status of controls, family history, sample size, and HWE in controls. Heterogeneity among studies was evaluated using Q test and I2 statistics. Fixed effect model (Mantel-Haenszel method) was used to calculate OR and 95% CI when P value of Q test was more than 0.10 or I2 value was less than 50%; otherwise, random effect model (DerSimonian-Laird method) was used. When the meta-analysis included 10 studies or more, publication bias was estimated using the visualizing Begg’s funnel plot, in which the log(OR) and its standard error of each study were indicated as Y- and X- axes respectively. An asymmetric funnel plot implied a possible publication bias. Furthermore, Egger’s linear regression test was utilized to determine the significance of asymmetry (P < 0.05 was considered to represent significant publication bias). Sensitivity analysis was performed with one study omitted at each time.
All analyses were performed using Stata 12.0, and two-sided tests with P value less than 0.05 was considered statistically significant unless otherwise specified.
Trial sequential analysis
Because of sparse data and repeated significance testing, meta-analyses may lead to type I error for the presence of systematic errors (bias) or random errors (play of chance) [25,26,27]. To assess our meta-analysis comprehensively, we performed TSA using the novel TSA software [28] to calculate the required information size (sample size) with an adjusted significance level. Briefly, we calculated the required information size on the basis of an overall type I error of 5%, an overall type II error of 20% (a power of 80%), and a relative risk reduction of 20%. Two-sided graphs were plotted using dotted black lines indicating boundaries for significance in a conventional meta-analysis, blue line indicating the cumulative Z-score, and red lines sloping inwards indicating trial sequential monitoring boundaries using adjusted P values.
Results
Study characteristics
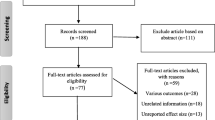
After strict screening, 34 eligible studies with 12,879 cases and 18,054 controls were identified in our meta-analysis (Fig. 1). In studies of Xu L et al. [24], Tommiska J et al. [29], and Akulevich NM et al. [30], two independent case-control studies were presented respectively; thus, each study was treated separately in our meta-analysis. For study of Xu L et al. [24], two parts of controls (HBC and PBC) were included; for study of Tommiska J et al. [29], two parts of cases (familial and unselected cases) were included; for study of Akulevich NM et al. [30], based on the condition of ionizing radiation (IR)-exposed or not, two separate studies were included, namely IR-induced papillary thyroid cancers (PTCs) vs IR-exposed controls, and sporadic PTCs vs non-exposed controls. Finally, 37 studies were included in the following analyses: 14 studies concentrated on effect of rs1801516 polymorphism on breast cancer risk [17, 18, 20, 22, 23, 29, 31,32,33,34,35,36,37], nine on thyroid cancer risk [19, 24, 30, 38,39,40,41], three on cervical cancer risk [42,43,44], two on colorectal cancer risk [45, 46], two on lung cancer risk [21, 47], one on bladder cancer risk [48], one on head and neck cancer risk [49], one on malignant melanoma risk [50], one on ovarian cancer risk [51], one on pancreatic cancer risk [52], one on prostate cancer risk [53], and one on renal cell cancer risk [54], respectively. Main characteristics of these studies are shown in Table 1 and Additional file 1: Table S1. Region-specified population was defined geographically in the 37 studies, 19 of which was European, 12 of which was North American, two of which was South American, three of which was Asian, and one of which was Oceanian. Cases in seven studies had a family history, and cases in the other 30 studies were unselected. Controls in 14 studies were HBC, controls in 17 studies were PBC, and six studies didn’t report the source of controls. A total of 25 studies had controls matched to cases for different factors; whereas, 12 studies had controls not matched to cases in that the controls were randomly selected. Genotyping methods were diverse, including real time polymerase chain reaction (RT-PCR), PCR-restriction fragment length polymorphism (PCR-RFLP), TaqManSNP (TaqMan), direct sequencing, microarray, and ten other methods. NOS scores of the included studies ranged from six to nine, indicating that the quality of studies in our meta-analysis is high.

Flow chart of the process of study identification and selection
HWE in controls and MAF in cases and controls for each study were obtained after reading the full text or calculated according to the genotype data (Table 2). As a result, rs1801516 genotype distribution of controls was in HWE for 30 studies, and was not in HWE for four studies; besides, genotype distribution could not be obtained for three studies. Therefore, to assess the potential influence of HWE on the overall results, subgroup analysis by HWE in controls was performed. For study of Calderon-Zuniga Fdel C et al., the minor allele A was not detected in controls.
Main results of meta-analyses
The pooled and subgroup meta-analyses of associations between rs1801516 polymorphism and cancer susceptibility are shown in Table 3. Overall, no significant association was found under any model of inheritance (AA vs GG + GA: OR = 0.91, 95% CI, 0.78–1.07; AA+GA vs GG: OR = 1.00, 95% CI, 0.90–1.11; AA vs GG: OR = 0.89, 95% CI, 0.75–1.06; GA vs GG: OR = 1.01, 95% CI, 0.91–1.13; GG + AA vs GA: OR = 1.00, 95% CI, 0.88–1.10). In subgroup analyses by region-specified population, significant associations were found in European (AA vs GG + GA: OR = 0.79, 95% CI, 0.65–0.96, P = 0.017; AA vs GG: OR = 0.79, 95% CI, 0.65–0.96, P = 0.017), South American (AA+GA vs GG: OR = 2.15, 95% CI, 1.37–3.38, P = 0.001; GA vs GG: OR = 2.19, 95% CI, 1.38–3.47, P = 0.001; GG + AA vs GA: OR = 0.46, 95% CI, 0.29–0.72, P = 0.001), and Asian (AA vs GG + GA: OR = 7.45, 95% CI, 1.31–42.46, P = 0.024; AA vs GG: OR = 7.40, 95% CI, 1.30–42.19, P = 0.024). In subgroup analyses by cancer types, significant decreased risk of breast cancer was found for those carrying AA genotype (AA vs GG: OR = 0.76, 95% CI, 0.59–0.98, P = 0.035). In subgroup analyses by family history, AA carriers had a significant decreased risk compared with GG carriers in those with family history (AA vs GG: OR = 0.68, 95% CI, 0.47–0.98, P = 0.039), and a borderline significance was found for AA vs GG + GA (OR = 0.70, 95% CI, 0.48–1.00, P = 0.051).
Heterogeneity analysis
We applied Q test and I2 statistics to evaluate the heterogeneity of our meta-analysis. Our results showed significant heterogeneity among studies for AA+GA vs GG (I2 = 60.8%, P < 0.001), GA vs GG (I2 = 77.1%, P < 0.001), and GG + AA vs GA (I2 = 74.4%, P < 0.001) models (Table 3). To further investigate the source of heterogeneity, we performed meta-regression analysis by region-specified population, cancer type, source of controls, matched controls or not, family history, sample size, and HWE in controls. As a result, family history was a source of heterogeneity for AA+GA vs GG (P = 0.040, 59% CI, 0.204–7.804) and GA vs GG (P = 0.044, 59% CI, 0.113–8.055), suggesting that family history may explain the among-studies’ heterogeneity under these two models. However, no factor was detected as a source of heterogeneity for GG + AA vs GA (Table 4).
Publication bias
Begg’s funnel plot and Egger’s linear regression test were used to assess the publication bias of studies in our meta-analysis. The shape of the funnel plots under four models seemed symmetrical (Fig. 2), and the results of Egger’s test revealed no evidence of significant publication bias (AA vs GG + GA: P = 0.266; AA+GA vs GG: P = 0.505; AA vs GG: P = 0.201; GA vs GG: P = 0.574; GG + AA vs GA: P = 0.587).

Funnel plots for publication bias of the meta-analysis on rs1801516 polymorphism and overall cancer risk. a recessive model: AA vs GG + GA; b dominant model: AA+GA vs GG; c codominant model: AA vs GG; d codominant model: GA vs GG; e overdominant model: AA+GG vs GA
Sensitivity analyses
We performed sensitivity analysis by excluding one study at each time to evaluate the influence of each individual study on the overall ORs and 95% CIs. The results showed that the pooled ORs and 95% CIs under any model of inheritance were not substantially altered after omitting any individual study (Fig. 3), suggesting that the results of our meta-analysis are credible.

Sensitivity analyses of the studies. a recessive model: AA vs GG + GA; b dominant model: AA+GA vs GG; c codominant model: AA vs GG; d codominant model: GA vs GG; e overdominant model: AA+GG vs GA
Trial sequential analysis
The results of TSA under four models (five comparisons) are shown in Fig. 4, and they were consistent with the results of the conventional meta-analyses. The blue lines of cumulative Z-score didn’t cross the trial sequential monitoring boundaries (red lines sloping inwards), suggesting there is no significant association between rs1801516 polymorphism and cancer risk. Moreover, sample sizes in our overall meta-analyses were all more than the required information sizes (AA vs GG + GA: 6429; AA+GA vs GG: 20201; AA vs GG: 8219; GA vs GG: 19885; GG + AA vs GA: 19209), suggesting that the results of our meta-analyses are reliable.

Trial sequential analysis of the association between rs1801516 polymorphism and overall cancer risk. The required information size was calculated based on a two side α = 5%, β = 25% (power 80%), and a relative risk reduction of 20%. a recessive model: AA vs GG + GA; b dominant model: AA+GA vs GG; c codominant model: AA vs GG; d codominant model: GA vs GG; e overdominant model: AA+GG vs GA
Discussion
Studies of rs1801516 polymorphism on cancer risk have been performed for more than ten cancers in previous studies, and breast cancer and thyroid cancer are the two most studied ones. So far, three meta-analyses have been performed on association between rs1801516 polymorphism and breast cancer risk [11, 12, 14], and two meta-analysis have been performed on the association between rs1801516 polymorphism and thyroid cancer risk [10, 55]. Moreover, one meta-analysis focused on this polymorphism and cancer risk despite of cancer types, but it was stratified by the status of radiation exposure [13]. In our meta-analysis, we assessed the association between rs1801516 polymorphism and overall cancer risk for the first time. We found that no significant association existed under any model of inheritance in the overall analysis. Our result was consistent with the finding of the previous study on rs1801516 polymorphism and cancer risk in population without radiation exposure [13]. Therefore, rs1801516 polymorphism may be not associated with overall cancer risk.
In subgroup analyses by region-specified population, cancer types, and family history, significant associations were found for European, South American, Asian, breast cancer, and those with family history. Firstly, results of subgroup analysis by region-specified population were interesting. In European and Asian, reversed results were observed for AA vs GG + GA and AA vs GG. The homozygote AA showed a protective effect against cancer in European, but it presented a susceptible effect for cancer in Asian. Therefore, rs1801516 polymorphism may exert inversed effect on European and Asian. In South American, the other three models (AA+GA vs GG, GA vs GG, and GG + AA vs GA) were significant. Susceptible effect for cancer was observed for AA+GA vs GG and GA vs GG, and protective effect against cancer was observed for GG + AA vs GA. We infer that the results in South American may be attributed to the heterozygote GA, which may be a risk genotype of cancer in South American. Populations from different region may be ethnically different, and this difference may in turn have an influence on cancer susceptibility. Studies have revealed cancer trends differed from ethnicity [56,57,58], and patients of different ethnicity presents different cancer phenotypes [59, 60]. Besides, discrepancy in distribution of rs1801516 genotype may exist in different populations. Secondly, subgroup analysis by cancer types in our study indicated that AA homozygotes have a relative low risk of breast cancer compared with GG carriers. Three previous meta-analyses [11, 12, 14] have been performed on association between rs1801516 polymorphism and breast cancer risk, and the result for AA vs GG + GA in study of Lu PH et al. [14] is significant, indicating that AA is a low risk genotype. Our results were consistent with those of Lu et al.. Moreover, 13 studies were included in our meta-analysis of breast cancer, which was much more than that in studies of Mao C et al.(eight studies included) [11], Gao LB et al. (nine studies included) [12], and Lu PH et al. (five studies included) [14]. Thus, compared with GG genotype, AA genotype of rs1801516 may be a potential protective factor of breast cancer. Thirdly, for those with family history, AA homozygotes presented low susceptibility of cancer in our meta-analysis. Impact of family history on cancer occurrence and clinical features has been found in different types of cancer, and family history may also exert an influence on cancer through interaction with gene polymorphism [61,62,63]. In Mao et al.’s meta-analysis [11], subgroup analysis was also performed by family history, but their results are not similar to ours. Difference in sample size between the two meta-analyses of ours and Mao et al.’s may result in the inconsistence of results.
In this present meta-analysis, heterogeneity was observed in models of AA+GA vs GG, GA vs GG, and GG + AA vs GA. To find the source of among-studies’ heterogeneity, we performed meta-regression analysis by region-specified population, cancer type, source of controls, matched controls or not, family history, sample size, and HWE in controls. As a result, family history was a source of heterogeneity for AA+GA vs GG and GA vs GG models. However, for GG + AA vs GA, none of the analyzed factors was detected as a source of heterogeneity. Lifestyle may be the source of heterogeneity. Lifestyle of the subjects, including smoking and alcohol consumption, influences on their susceptibility to cancer [64,65,66]. However, the 37 studies included in our meta-analysis do not provide adequate information on lifestyle. Moreover, genotyping methods of the included studies are various: more than ten methods used in all the included studies, and multiple methods were used in several individual studies. Diversity of genotyping methods may also be a reason of heterogeneity, but because of diverse methods, we do not put genotyping methods into the analysis of meta-regression. In addition, matching criteria of the included studies with controls matched to cases are different, giving rise to the heterogeneity possibly.
Meta-analysis may report false positive results for the risk of type I error, and such results are commonly attributed to publication bias, heterogeneity among studies, and low quality of the studies. However, a limited number of trials may not give enough information size, thereby leading to a false estimation [67]. In order to comprehensively evaluate the impact of ATM rs1801516 polymorphism on cancer risk, we performed TSA to reduce the risk of type I error and to estimate whether further studies are required by calculating the required information size. Sample size in our meta-analysis was more than the required information size, indicating that the results of our meta-analyses are reliable and sufficient to draw a conclusion.
We must admit that there are some limitations in our meta-analysis. Firstly, because of the difference in data presentation of age between studies (mean age, median age, and age group), we didn’t assess the risk stratified by age. Secondly, environmental factors and life style information were not available for all studies, thus effects of these variables were not taken into consideration. Thirdly, year of data collection may also have an effect on heterogeneity, but not all studies in our meta-analysis provide this information, thus year of data collection was not analyzed in our meta-analysis. Fourthly, 12 types of cancer were included in our meta-analysis. However, only one or two studies were performed on the cancers except breast cancer, thyroid cancer, and cervical cancer, and this may potentially make the result biased.
Conclusions
In summary, ATM rs1801516 polymorphism is not associated with overall cancer risk in total population. However, for subgroup analyses, rs1801516 polymorphism is especially associated with breast cancer risk; in addition, this polymorphism is associated with overall cancer risk in Europeans, South Americans, Asians, and those with family history. Owing to the limitations mentioned above, our results should be interpreted with caution.
Abbreviations
- A-T:
-
Ataxia-telangiectasia
- ATM:
-
Ataxia telangiectasia mutated
- CI:
-
Confidence interval
- DSB:
-
Double-strand break
- GWAS:
-
Genome-wide association studies
- HBC:
-
Hospital-based controls
- HWE:
-
Hardy-Weinberg equilibrium
- IR:
-
Ionizing radiation
- MAF:
-
Minor allele frequency
- NOS:
-
Newcastle-Ottawa Scale
- OR:
-
Odds ratio
- PBC:
-
Population-based controls
- PCR-RFLP:
-
PCR-restriction fragment length polymorphism
- PI3-K:
-
Phosphoinositide 3-kinase
- PRISMA:
-
Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- PTC:
-
Papillary thyroid cancer
- RT-PCR:
-
Real time polymerase chain reaction
- SNP:
-
Single nucleotide polymorphism
- TaqMan:
-
TaqManSNP
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29.
WHO. GLOBOCAN 2012. Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. http://globocan.iarc.fr. Accessed 17 May 2017.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268(5218):1749–53.
Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9(10):759–69.
Morrell D, Cromartie E, Swift M. Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst. 1986;77(1):89–92.
Swift M, Morrell D, Massey RB, Chase CL. Incidence of cancer in 161 families affected by ataxia-telangiectasia. N Engl J Med. 1991;325(26):1831–6.
Chaudhary MW, Al-Baradie RS. Ataxia-telangiectasia: future prospects. Appl Clin Genet. 2014;7:159–67.
Barrett JH, Iles MM, Harland M, Taylor JC, Aitken JF, Andresen PA, et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat Genet. 2011;43(11):1108–13.
Kang J, Deng XZ, Fan YB, Wu B. Relationships of FOXE1 and ATM genetic polymorphisms with papillary thyroid carcinoma risk: a meta-analysis. Tumour Biol. 2014;35(7):7085–96.
Mao C, Chung VCH, He BF, Luo RC, Tang JL. Association between ATM 5557G > a polymorphism and breast cancer risk: a meta-analysis. Mol Biol Rep. 2012;39(2):1113–8.
Gao LB, Pan XM, Sun H, Wang X, Rao L, Li LJ, et al. The association between ATM D1853N polymorphism and breast cancer susceptibility: a meta-analysis. J Exp Clin Cancer Res. 2010;29:117.
Zhao YG, Yang L, Wu D, He H, Wang MM, Ge TW, et al. Gene-environment interaction for polymorphisms in ataxia telangiectasia-mutated gene and radiation exposure in carcinogenesis: results from two literature-based meta-analyses of 27120 participants. Oncotarget. 2016;7(47):76867–81.
Lu PH, Wei MX, Si SP, Liu XA, Shen W, Tao GQ, et al. Association between polymorphisms of the ataxia telangiectasia mutated gene and breast cancer risk: evidence from the current studies. Breast Cancer Res Treat. 2011;126(1):141–8.
Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, Ioannidis JP, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med. 2009;6(7):e1000100.
The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses. http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm. Accessed 5 Aug 2017.
Sommer SS, Buzin CH, Jung M, Zheng J, Liu Q, Jeong SJ, et al. Elevated frequency of ATM gene missense mutations in breast cancer relative to ethnically matched controls. Cancer Genet Cytogenet. 2002;134(1):25–32.
Gonzalez-Hormazabal P, Bravo T, Blanco R, Valenzuela CY, Gomez F, Waugh E, et al. Association of common ATM variants with familial breast cancer in a south American population. BMC Cancer. 2008;8:117.
Maillard S, Damiola F, Clero E, Pertesi M, Robinot N, Rachedi F, et al. Common variants at 9q22.33, 14q13.3, and ATM loci, and risk of differentiated thyroid cancer in the French Polynesian population. PLoS One. 2015;10(4):e0123700.
Calderon-Zuniga Fdel C, Ocampo-Gomez G, Lopez-Marquez FC, Recio-Vega R, Serrano-Gallardo LB, Ruiz-Flores P. ATM polymorphisms IVS24-9delT, IVS38-8T>C, and 5557G>a in Mexican women with familial and/or early-onset breast cancer. Salud Publica Mex. 2014;56(2):206–12.
Yang H, Spitz MR, Stewart DJ, Lu C, Gorlov IP, Wu X. ATM sequence variants associate with susceptibility to non-small cell lung cancer. Int J Cancer. 2007;121(10):2254–9.
Bretsky P, Haiman CA, Gilad S, Yahalom J, Grossman A, Paglin S, et al. The relationship between twenty missense ATM variants and breast cancer risk: the multiethnic cohort. Cancer Epidemiol Biomark Prev. 2003;12(8):733–8.
Hirsch AE, Atencio DP, Rosenstein BS. Screening for ATM sequence alterations in African-American women diagnosed with breast cancer. Breast Cancer Res Treat. 2008;107(1):139–44.
Xu L, Morari EC, Wei Q, Sturgis EM, Ward LS. Functional variations in the ATM gene and susceptibility to differentiated thyroid carcinoma. J Clin Endocrinol Metab. 2012;97(6):1913–21.
Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta-analyses may be inconclusive-trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta-analyses. Int J Epidemiol. 2009;38(1):287–98.
Turner RM, Bird SM, Higgins JP. The impact of study size on meta-analyses: examination of underpowered studies in Cochrane reviews. PLoS One. 2013;8(3):e59202.
Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta-analysis. J Clin Epidemiol. 2008;61(1):64–75.
TSA - Trial Sequential Analysis. http://www.ctu.dk/tsa/. Accessed 15 Oct 2017.
Tommiska J, Jansen L, Kilpivaara O, Edvardsen H, Kristensen V, Tamminen A, et al. ATM variants and cancer risk in breast cancer patients from southern Finland. BMC Cancer. 2006;6:209.
Akulevich NM, Saenko VA, Rogounovitch TI, Drozd VM, Lushnikov EF, Ivanov VK, et al. Polymorphisms of DNA damage response genes in radiation-related and sporadic papillary thyroid carcinoma. Endocr Relat Cancer. 2009;16(2):491–503.
Tapia T, Sanchez A, Vallejos M, Alvarez C, Moraga M, Smalley S, et al. ATM allelic variants associated to hereditary breast cancer in 94 Chilean women: susceptibility or ethnic influences? Breast Cancer Res Treat. 2008;107(2):281–8.
Schrauder M, Frank S, Strissel PL, Lux MP, Bani MR, Rauh C, et al. Single nucleotide polymorphism D1853N of the ATM gene may alter the risk for breast cancer. J Cancer Res Clin Oncol. 2008;134(8):873–82.
Angele S, Romestaing P, Moullan N, Vuillaume M, Chapot B, Friesen M, et al. ATM haplotypes and cellular response to DNA damage: association with breast cancer risk and clinical radiosensitivity. Cancer Res. 2003;63(24):8717–25.
Dork T, Bendix R, Bremer M, Rades D, Klopper K, Nicke M, et al. Spectrum of ATM gene mutations in a hospital-based series of unselected breast cancer patients. Cancer Res. 2001;61(20):7608–15.
Heikkinen K, Rapakko K, Karppinen SM, Erkko H, Nieminen P, Winqvist R. Association of common ATM polymorphism with bilateral breast cancer. Int J Cancer. 2005;116(1):69–72.
Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38(8):873–5.
Buchholz TA, Weil MM, Ashorn CL, Strom EA, Sigurdson A, Bondy M, et al. A Ser49Cys variant in the ataxia telangiectasia, mutated, gene that is more common in patients with breast carcinoma compared with population controls. Cancer. 2004;100(7):1345–51.
Wojcicka A, Czetwertynska M, Swierniak M, Dlugosinska J, Maciag M, Czajka A, et al. Variants in the ATM-CHEK2-BRCA1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosomes Cancer. 2014;53(6):516–23.
Damiola F, Byrnes G, Moissonnier M, Pertesi M, Deltour I, Fillon A, et al. Contribution of ATM and FOXE1 (TTF2) to risk of papillary thyroid carcinoma in Belarusian children exposed to radiation. Int J Cancer. 2014;134(7):1659–68.
Halkova T, Dvorakova S, Sykorova V, Vaclavikova E, Vcelak J, Vlcek P, et al. Polymorphisms in selected DNA repair genes and cell cycle regulating genes involved in the risk of papillary thyroid carcinoma. Cancer Biomark. 2016;17(1):97–106.
Pereda CM, Lesueur F, Pertesi M, Robinot N, Lence-Anta JJ, Turcios S, et al. Common variants at the 9q22.33, 14q13.3 and ATM loci, and risk of differentiated thyroid cancer in the Cuban population. Bmc Genetics. 2015;16:22.
Alsbeih G, Al-Harbi N, El-Sebaie M, Al-Badawi I. HPV prevalence and genetic predisposition to cervical cancer in Saudi Arabia. Infect Agent Cancer. 2013;8(1):15.
Oliveira S, Ribeiro J, Sousa H, Pinto D, Baldaque I, Medeiros R. Genetic polymorphisms and cervical cancer development: ATM G5557A and p53bp1 C1236G. Oncol Rep. 2012;27(4):1188–92.
Al-Harbi NM, Bin Judia SS, Mishra KN, Shoukri MM, Alsbeih GA. Genetic predisposition to cervical Cancer and the association with XRCC1 and TGFB1 polymorphisms. Int J Gynecol Cancer. 2017;27(9):1949–56.
Maillet P, Chappuis PO, Vaudan G, Dobbie Z, Muller H, Hutter P, et al. A polymorphism in the ATM gene modulates the penetrance of hereditary non-polyposis colorectal cancer. Int J Cancer. 2000;88(6):928–31.
Kristensen AT, Bjorheim J, Wiig J, Giercksky KE, Ekstrom PO. DNA variants in the ATM gene are not associated with sporadic rectal cancer in a Norwegian population-based study. Int J Color Dis. 2004;19(1):49–54.
Landi S, Gemignani F, Canzian F, Gaborieau V, Barale R, Landi D, et al. DNA repair and cell cycle control genes and the risk of young-onset lung cancer. Cancer Res. 2006;66(22):11062–9.
Wu X, Gu J, Grossman HB, Amos CI, Etzel C, Huang M, et al. Bladder cancer predisposition: a multigenic approach to DNA-repair and cell-cycle-control genes. Am J Hum Genet. 2006;78(3):464–79.
Al-Hadyan KS, Al-Harbi NM, Al-Qahtani SS, Alsbeih GA. Involvement of single-nucleotide polymorphisms in predisposition to head and neck cancer in Saudi Arabia. Genet Test Mol Biomarkers. 2012;16(2):95–101.
Pena-Chilet M, Blanquer-Maceiras M, Ibarrola-Villava M, Martinez-Cadenas C, Martin-Gonzalez M, Gomez-Fernandez C, et al. Genetic variants in PARP1 (rs3219090) and IRF4 (rs12203592) genes associated with melanoma susceptibility in a Spanish population. BMC Cancer. 2013;13:160.
Tecza K, Pamula-Pilat J, Kolosza Z, Radlak N, Grzybowska E. Genetic polymorphisms and gene-dosage effect in ovarian cancer risk and response to paclitaxel/cisplatin chemotherapy. J Exp Clin Cancer Res. 2015;34:2.
Li D, Suzuki H, Liu B, Morris J, Liu J, Okazaki T, et al. DNA repair gene polymorphisms and risk of pancreatic cancer. Clin Cancer Res. 2009;15(2):740–6.
Angele S, Falconer A, Edwards SM, Dork T, Bremer M, Moullan N, et al. ATM polymorphisms as risk factors for prostate cancer development. Br J Cancer. 2004;91(4):783–7.
Margulis V, Lin J, Yang H, Wang W, Wood CG, Wu X. Genetic susceptibility to renal cell carcinoma: the role of DNA double-strand break repair pathway. Cancer Epidemiol Biomark Prev. 2008;17(9):2366–73.
Yu H, Duan YQ, Zhang C, Zhang JR. Association between ATM polymorphism and the risk of thyroid cancer: a meta-analysis. Int J Clin Exp Med. 2017;10(10):14869–75.
Singh E, Joffe M, Cubasch H, Ruff P, Norris SA, Pisa PT. Breast cancer trends differ by ethnicity: a report from the south African National Cancer Registry (1994-2009). Eur J Pub Health. 2017;27(1):173–8.
Byun JS, Park S, Caban A, Jones A, Gardner K. Linking Race, Cancer Outcomes and Tissue Repair. Am J Pathol. 2018;188(2):317–28.
Friedrich P, Itriago E, Rodriguez-Galindo C, Ribeiro K. Racial and Ethnic Disparities in the Incidence of Pediatric Extracranial Embryonal Tumors. J Natl Cancer Inst. 2017;109(10):djx050.
Askari A, Nachiappan S, Currie A, Latchford A, Stebbing J, Bottle A, et al. The relationship between ethnicity, social deprivation and late presentation of colorectal cancer. Cancer Epidemiol. 2017;47:88–93.
Wu VJ, Pang D, Tang WW, Zhang X, Li L, You Z. Obesity, age, ethnicity, and clinical features of prostate cancer patients. Am J Clin Exp Urol. 2017;5(1):1–9.
Poynter JN, Richardson M, Roesler M, Krailo M, Amatruda JF, Frazier AL. Family history of cancer in children and adolescents with germ cell tumours: a report from the Children's oncology group. Br J Cancer. 2018;118(1):121–6.
Hwang M, Park B. Association between health behaviors and family history of Cancer in Cancer survivors: data from the Korean genome and epidemiology study. J Cancer Prev. 2017;22(3):166–73.
Fagerholm R, Faltinova M, Aaltonen K, Aittomaki K, Heikkila P, Halttunen-Nieminen M, et al. Family history influences the tumor characteristics and prognosis of breast cancers developing during postmenopausal hormone therapy. Fam Cancer. 2018;17(3):321–31.
Osazuwa-Peters N, Adjei Boakye E, Chen BY, Tobo BB, Varvares MA. Association Between Head and Neck Squamous Cell Carcinoma Survival, Smoking at Diagnosis, and Marital Status. JAMA Otolaryngol Head Neck Surg. 2017. Epub ahead of print.
Bekalu MA, Minsky S, Viswanath K. Beliefs about smoking-related lung cancer risk among low socioeconomic individuals: the role of smoking experience and interpersonal communication. Glob Health Promot. 2017;1757975917732758. Epub ahead of print.
Garcia Lavandeira JA, Ruano-Ravina A, Kelsey KT, Torres-Duran M, Parente-Lamelas I, Leiro-Fernandez V, et al. Alcohol consumption and lung cancer risk in never smokers: a pooled analysis of case-control studies. Eur J Pub Health. 2018;28(3):521–7.
Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, et al. Can trial sequential monitoring boundaries reduce spurious inferences from meta-analyses? Int J Epidemiol. 2009;38(1):276–86.
Acknowledgements
The authors would like to thank Dr. Charkos Tesfaye Getachew and Dr. Oumer Kemal Sherefa for their critical reading of this manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81602907 and 81702606), the funds from Science and Technology Commission of Jilin Province (20170520007JH), and the funds from Commission of Health and Family Planning of Jilin Province (2017Q037). The National Natural Science Foundation of China plays role in study design and in writing the manuscript, the others play role in collection, analysis, and interpretation of data.
Availability of data and materials
The datasets analyzed during the current study are available in the PubMed, Web of Science, and Embase repositories. Persistent web links of each study included in the datasets are provided in the “Reference” part.
Author information
Authors and Affiliations
Contributions
YG, YC and YL conceived and designed the study; YG, JS, SQ, YQ and XZ performed study selection, data extraction, statistical analysis and interpretation; JS and SQ performed quality control of data; YG wrote the manuscript; YQ, YC and YL revised the manuscript. All authors approved the final manuscript for submission and publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Matching criteria and genotyping method of the eligible studies included in the meta-analysis. (DOCX 24 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gu, Y., Shi, J., Qiu, S. et al. Association between ATM rs1801516 polymorphism and cancer susceptibility: a meta-analysis involving 12,879 cases and 18,054 controls. BMC Cancer 18, 1060 (2018). https://doi.org/10.1186/s12885-018-4941-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-018-4941-1