
Abstract
Background
Giant Cell Tumour of Bone (GCT) is a locally aggressive primary bone tumour that usually occurs at the epiphyses of the long bones of the appendicular skeleton with a tendency to recurrence. Recurrent somatic H3F3A mutations have been described in 92% of GCT cases. GCTs involving the Clivus are extremely rare lesions and less than 15 cases are described in the literature. They represent a surgery challenge and are easily misdiagnosed. Our aim was to reveal if the genetic bases underlying Clival GCTs were the same of GCTs of long bones to improve the diagnosis and treatment.
Methods
The targeted somatic sequencing of GCT-related genes (H3F3A, H3F3B, IDH1, IDH2 and ZNF687) was performed on Clival GCT biopsies of two different cases. Histological analyses on the same tissues were used to detect the neoplastic population and its expression profile.
Results
Sanger sequencing revealed that both patients were positive for the p.Gly34Trp mutation in the H3F3A gene. Immunofluorescence assay using monoclonal antibody, specifically detecting the mutant H3.3, highlighted that the mutation only involved the mononuclear cell population and not the multinucleated giant cells. Moreover, immunohistochemistry assay showed that RANKL was highly expressed by the stromal cells within Clival GCT, mimicking what happens in GCT of the long bones. In addition, systematic literature review allowed us to generate a histology-based diagnostic algorithm of the most common clival lesions.
Conclusions
We conclude that the Clival GCT is genetically defined by somatic mutation in the H3F3A gene, linking it to the GCT of long bones. The similarity with GCTs of long bones let us to hypothesize the utility of Denosumab therapy (already effective for GCTs) in these surgically challenging cases. Moreover, H3F3A genetic screening can be combined to the histological analysis to differentiate GCTs from morphologically similar giant cell-rich sarcomas, while the histological diagnostic algorithm could help the differential diagnosis of other clival lesions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Giant cell tumour of bone (GCT) is a primary intramedullary neoplasm that accounts for 5% of skeletal tumours, composed by numerous multinucleated osteoclast-like giant cells evenly scattered throughout the mass and ovoid or spindle mononuclear stromal cells [1]. GCT is generally considered a benign tumour, even though it is characterised by localised bone destruction due to the osteolytic properties of osteoclast-like giant cells that express the markers involved in bone resorption activity [2]. Although the giant cells are a significant part of this tumour, the stromal cells constitute the actual neoplastic component. Indeed, Behjati et al. recently described recurrent driver somatic mutations in the H3F3A gene only restricted to the stromal cell population, rather than to cells of the osteoclast lineage, demonstrating that GCT is a mesenchymal neoplasm [3]. Particularly, an exquisite specificity for H3.3 alterations was observed among different bone tumours, emphasizing the importance of genotyping tumours for diagnostic purposes [3,4,5]. On the contrary, we recently highlighted that giant cell tumour, when arising on Paget’s disease of bone – a disorder of bone remodelling – shows a different genetic signature characterised by a germline mutation in the ZNF687 gene [6, 7].
Even though giant cell tumour shows a low potential for metastasis, it may locally recur at high rate [8]. Therefore, a complete resection accompanied by adjuvant therapy to prevent tumour recurrence is required. Denosumab has been demonstrated as an effective therapy in GCT for tumour control. This fully humanised monoclonal antibody selectively targets RANKL, thus inhibiting its interaction with RANK receptor on the surface of osteoclast precursors and preventing bone destruction activity [9]. Denosumab treatment in GCT has been shown to effectively reduce not only the number of giant cells but also the relative content of proliferative stromal cells, promoting new bone formation [9, 10].
GCT typically occurs when the growth plate has closed and therefore is frequently observed in skeletally mature individuals with its peak incidence in the third and fourth decade of life [11]. The majority of GCTs develop as single lesions and are located at the epiphyses of long bones, predominantly affecting the distal femur, the proximal tibia, the distal radius and the proximal humerus [12, 13]. GCTs involving other anatomic sites are uncommon and only less than 1% of all reported GCTs occurs in the skull, where they preferentially affect the sphenoid and temporal bone [14]. Specifically, primary giant cell tumours of the clivus are extremely rare lesions, with less than 15 cases described in the literature, that typically present with compression of the cranial nerves and consequent diplopia, headache and deafness [15,16,17,18,19,20,21,22,23,24,25,26]. Albeit histologically benign, Clival GCT can be clinically devastating because of its anatomical location and destruction of vital structures [15]. The tumour also shows a high tendency to local recurrence, thus making the total surgical resection essential [15, 19]. However, the complete removal is not always feasible and an adjuvant treatment (chemotherapy or radiotherapy) is often used [15,16,17, 20, 24].
In the present article, we defined the genetic basis of giant cell tumour of the clivus, demonstrating the presence of somatic mutation in the H3F3A gene in tumour biopsies of two patients, highlighting that Clival GCT can be considered like GCT of long bones.
Methods
Patients and tissues
The patient material comprises primary giant cell tumours of the clivus. Tissue samples were obtained as Formalin Fixed Paraffin Embedded (FFPE) specimens from 2 patients surgically treated in Università Vita-Salute San Raffaele, Milan, Italy (patient 1) and Department of Neurosurgery, Umberto I General Hospital, Ancona, Italy (patient 2).
DNA extraction
Sections (7-μm-thick) were cut from FFPE tissue blocks and subjected to DNA extraction with GeneRead DNA FFPE Kit (Qiagen), following the manufacturer’s instructions.
Genetic screening
Mutation analysis of GCT-related genes (H3F3A, H3F3B, IDH1, IDH2, and ZNF687) was conducted by PCR followed by Sanger sequencing, as recently reported [7].
Allele-specific sequencing
The molecular cloning of H3F3A DNA sequence containing c.G103T mutation was carried out as previously described [7]. Briefly, H3F3A genomic region was amplified as described above, subcloned into pJET1.2/blunt cloning vector (Thermo Fisher Scientific), and subjected to Sanger sequencing.
Immunohistochemistry and immunofluorescence analysis
Tumour sections (7-μm-thick) of paraffin blocks were cut using a microtome and collected on Superfrost glass slides (Thermo Fisher Scientific). FFPE tumour tissues were then deparaffinised and rehydrated as we recently reported [7]. Sections were incubated with primary antibodies for rabbit monoclonal anti-H3.3 p.Gly34Trp (RevMab Biosciences, clone RM263, recently used by Lüke et al.), mouse monoclonal anti-TRAP (Thermo Fisher Scientific MA5–12387), mouse monoclonal anti-Tenascin C (Abcam ab6393) and rabbit polyclonal anti-RANKL (Abcam ab9957) [27]. Immunofluorescence sections were observed using a Nikon’s A1R confocal laser microscope, while immunohistochemistry sections were analysed using Nikon Intensilight C-HGFI.
Results
Clinical case description
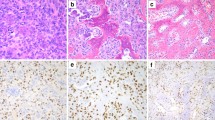
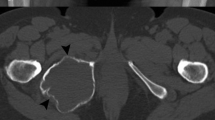
We recently recruited a 55-year-old female (hereafter referred to as “patient 1”), diagnosed in 2014 with giant cell tumour of the clivus. Since 2013, she complained holocranial headache associated with vomiting, tongue numbness and difficulty with swallowing and speech. Computed tomography (CT) showed a lytic mass 5 cm in size involving the clivus. Magnetic resonance imaging (MRI) revealed a lobulated mass (5 × 2,8 × 3,8 cm) originating from the clivus and extending into the sella and the epistropheus. In 2014, she underwent surgical removal of the mass through suboccipital approach and from the postoperative histopathology the diagnosis of Clival GCT was made. The tumour was highly vascularized and composed of numerous osteoclast-like giant cells that were bigger than those usually described in GCTs, with 50 to 70 nuclei, scattered on a background of mononuclear cells (Fig. 1a). However, one month after the surgical intervention, the tumour mass grew, symptoms presented again and the mass was newly removed. Postoperatively, the patient received tomotherapy against the residual lesion but only dysphonia improved. Nevertheless, so far in 2016 she suffers of worsening headache, tongue paralysis and dysphagia. MRI demonstrated that a recurrence occurred and showed growth of the tumour (5,3 × 2,1 × 3,9 cm), with compression of hypoglossal nerve (Fig. 2). She has been planned for resurgery.

Case presentation. a Histology of the specimen excised in 2014 from patient 1, showing numerous multinucleated osteoclast-like giant cells scattered in a background of mononuclear cells (hematoxylin and eosin stain; 20× objective). b Haematoxylin and eosin staining of tumor biopsy of patient two. Smaller osteoclast-like giant cells are observable (20× objective)

MRI images. Sagittal, axial and coronal magnetic resonance images of patient 1, depicting the large and recurrent giant cell tumour originating from the clivus (indicated by the red arrow in the sagittal view), with a C1 vertebral infiltration
In 2013, the patient 2 was operated through endoscopic endonasal approach to remove the giant cell tumour of the clivus, responsible for his progressive headache and diplopia. After surgery, the patient was asymptomatic and did not undergo radiation therapy. Brain MRI, regularly performed up to six years after the operation, revealed that he did not present any relapse of the disease [18]. The haematoxylin and eosin staining that we performed on tumour sample confirmed the clinical diagnosis, even though the size of giant cells was remarkably smaller than in patient 1 (Fig. 1b).
H3F3A somatic mutation defines the molecular basis of Clival GCT
The molecular bases underlying Clival GCT pathogenesis have never been investigated, likely because of the extreme rarity of this skull tumour. Here, in an attempt to prove that GCT of the clivus may be caused by the same genetic alteration of GCT of long bones, we performed the molecular analysis of the GCT-related genes (H3F3A, H3F3B, IDH1 and IDH2) as well as of the ZNF687 gene that we recently identified as gene responsible for giant cell tumour associated with Paget’s disease of bone [3, 6, 28]. We analysed the tumour specimens of the two patients described above and revealed that both harboured the heterozygous p.Gly34Trp mutation in the H3F3A gene; whereas the coding regions of the other analysed genes resulted negative for any mutations (Fig. 3a). Moreover, we performed the genetic screening on DNA extracted from peripheral blood of patient 1 and, as expected, we did not detect the H3F3A alteration, thus confirming that the mutation was somatically acquired. Given the somatic origin of the mutations, Sanger sequencing detected a very low peak in correspondence of the investigated nucleotide in both patients, making the signal from the mutations hardly distinguishable from background noise. Therefore, we cloned both DNA fragments carrying the p.Gly34Trp mutation and, as shown in Fig. 3b, we confirmed the presence of the mutant allele.

H3F3A mutation causes GCT of the clivus. a Sequence electropherograms of the PCR product of the H3F3A gene, showing the low mutational peak at position c.103 in patient 1 (left) and patient 2 (right). Asterisks denote the mutated base. b Representative electropherogram of patient 1 showing the allele specific sequencing of cloned DNA fragment and demonstrating the presence of p.Gly34Trp mutation in H3F3A. c Immunofluorescent visualization of the H3F3A mutation in the nuclei of mononuclear cells, while osteoclastic giant cells are negative and only react with TRAP antibody (20× objective, scale bar 100 μm). Note the higher number of neoplastic cells in Clival GCT of patient 1 (left) compared to patient 2 (right)
Stromal cells are the malignant component of Clival GCT and express high levels of tenascin-C
Considering that our samples of Clival GCT showed a different clinical aggressiveness of the tumour, we aimed at defining the amount of neoplastic cells within the mass through immunofluorescence assay, using a mutation-specific monoclonal antibody. We demonstrated that the p.Gly34Trp mutation in H3F3A disturbs the mononuclear cells that showed a strong nuclear staining in the tumour biopsies of both patients, whereas, as expected, the nuclei of all TRAP-positive giant cells were negative, indicating that the mutation only regarded the mononuclear cell compartment (Fig. 3c). This analysis allowed us to observe that the number of malignant cells was higher in the tumour biopsy of patient 1, correlating with her more severe phenotype.
Given the significant expression of the glycoprotein Tenascin-C in GCT patients and its association with local relapse, we also evaluated its immunohistochemical expression in both tumour biopsies. Interestingly, we found strong immunopositivity also in these two Clival GCTs, with a reticulate organization pattern in the extracellular matrix (Fig. 4). This result is in agreement with the local recurrence of the tumor in patient 1, occurred two years after the surgical operation.

Clival GCT expresses Tenascin-C and high levels of RANKL. Tenascin-C reactivity in the extracellular matrix and RANKL-positive mononuclear cells in tumour biopsies of patient 1 (left) and patient 2 (right) are shown (20× objective, scale bar 100 μm)
Clival GCTs express high levels of RANK-ligand mimicking GCT of long bones
It is well known that GCTs of long bones express high levels of RANKL, justifying the introduction of the monoclonal antibody Denosumab in GCT clinical practice [9]. To reveal if Clival GCT and GCT of long bones shared a similar expression pattern, we also evaluated RANKL levels in the Clival GCT biopsies of both patients. Immunohistochemistry assay detected a high number of RANKL-positive cells, showing an expression pattern similar to that recently reported for GCT of long bones by us and others (Fig. 4) [7]. This result led us to speculate that Denosumab treatment could represent an effective therapy for this surgically challenging tumour.
Systematic literature review confirms that skull GCTs are rare entities
Giant cell tumours seldom occur in the skull. Reviewing data from three extensive studies in the literature, we were able to estimate that the frequency of GCTs arising from the calvarial bones is 0,51% of all GCTs. In 1985, among a collection of 407 cases of GCT, David Dahlin highlighted only 4 tumours occurring in the skull [29]. Subsequently, Bertoni et al. reviewed 2046 GCT cases (546 of which were contained in the Mayo Clinic files) and found only 15 cases affecting the skull bones [14]. In the Rizzoli Case Archive, among 1449 GCT cases collected from 1900 to 2012, only 1 was located at skull [30]. Therefore, skull GCTs have been observed only in 20 out of 3902 GCT patients.
Moreover, we analysed 104 case reports of GCTs of the skull base from 1969 to 2017 and highlighted that the sphenoid and temporal bones were those preferentially involved by GCT, with a frequency of 47% and 28%, respectively [14, 29, 31,32,33,34,35,36,37,38,39,40,41,42,43,44,45]. Clivus as primary site of GCT was described in 12 out of 104 reports (12%), while occipital and frontal GCTs showed an incidence of 9% and 4%, respectively (Fig. 5) [15,16,17,18,19,20,21,22,23,24,25,26, 38, 46,47,48,49,50,51].

Diagrammatic representation of the human skull in sagittal section, identifying the main bones and cavity. The location of lesions in 104 cases of skull GCTs is indicated by arrows. Adapted from Anatomy of the Human Body [77]
Histological diagnostic algorithm permits the differential diagnosis of the main clival lesions
Giant cell tumour is among the rarest lesions arising within the clivus but pathologies of the clivus are represented by a wide range of diseases, and hence the differential diagnosis can include: chordoma; chondrosarcoma; meningioma; osteosarcoma; pituitary adenoma; lymphoma; and plasmacytoma. These tumours have similar anatomical characteristics and imaging approaches are not sufficient to distinguish them. However, differentiating among these masses is essential to define the proper pharmacological treatment. Here, we generated a diagnostic algorithm for the most frequent lesions, showing in Table 1 their gross and microscopic appearance as well as their distinctive immunostaining pattern.
Chordomas are the most common pathologies in the skull, accounting for 42% of all chordomas, and usually occur in the vicinity of the clivus [52, 53]. They arise from notochord remnants and, although slow-growing, they are locally aggressive and tend to recur after surgical removal, behaving like malignant tumours [52]. Chondrosarcomas comprise 6% of skull base tumours and develop from the transformation of mesenchymal cells producing cartilage. They show a high predilection for the petroclival synchondrosis, and yet they can also have a midline skull base location [54, 55]. Therefore, chordomas and chondrosarcomas may occupy the same anatomic location, show the same clinical symptoms (headache and diplopia) and a similar MRI appearance (hypointensity on T1 and hyperintensity on T2), and indeed the two tumours are frequently confused with one another [56]. Nevertheless, chordomas show a substantially worse prognosis and positivity to the markers EMA, cytokeratin and brachyury helps to distinguish it from chondrosarcoma [57] (Table 1).
Meningiomas are tumours originating from the arachnoidal cells and, being the clivus covered with dura mater, it can be affected. Specifically, meningiomas more frequently arise in the petroclival junction. Patients commonly present with headache, seizures and hearing disturbance. This tumour has a dual mesenchymal and epithelial differentiation potential, and hence shows positivity to both vimentin and keratin [58, 59]. Of note, keratin expression can be observed in 75% of malignant meningiomas; whereas its expression is never detected in benign meningiomas [60] (Table 1).
Osteosarcomas are highly aggressive lesions caused by osteoid-producing malignant cells. They primarily affect the metaphysis of long bones but 6–10% of them may take place within the skull [61]. Being a tumour of mesenchymal origin, osteosarcoma shows positivity to vimentin. However, the presence of osteoid as well as the positivity to alkaline phosphatase represent the criteria for the differential diagnosis of osteosarcomas [62].
Pituitary adenomas are common intracranial tumours, comprising about 10% of skull lesions. They frequently invade the adjacent structures, even though clival involvement is rarer. These lesions can be asymptomatic or can cause headache and amenorrhea [63].
Lymphomas and plasmacytomas are cancers of the immune system; lymphomas develop from lymphocytes, while plasmacytomas are monoclonal proliferation of plasma cells. The sites most commonly affected are bones with active bone marrow hematopoiesis and the skull is seldom involved [64, 65]. Their microscopic appearance is very similar but negative staining for markers such as CD45 and CD20 is useful in supporting the plasmacytoma as differential diagnosis [66] (Table 1).
On the other hand, clivus can also be the site of metastatic lesions, commonly arising from prostate carcinoma (18,1%), hepatocellular carcinoma (10,6%), and thyroid follicular carcinoma (8,5%). Although they are more uncommon than primary clival tumours, they should be considered in the differential diagnosis that should be made using the histological markers specific for the primary cancers [67,68,69].
Discussion
Giant cell tumours of the skull account for 0,51% of all GCTs and tend to affect the sphenoid and the temporal bone. The selective and preferential occurrence of the tumour in these bones rather than other calvarial bones may depend on their embryologic origin: as in the case of long bones, also the sphenoid and the temporal bone are generated through endochondral bone formation; whereas the other skull bones are produced by an intramembranous formation [14]. In fact, we pointed out that the occipital and the frontal bones are the cranial sites less frequently involved by GCT. The giant cell tumour of the clivus is also a very rare lesion, with only 12 cases described to date [15,16,17,18,19,20,21,22,23,24,25,26]. However, the incidence of the Clival GCT (12% of all skull GCTs) is not the lowest of all and this may be due to its location at the sphenooccipital synchondrosis, a joint composed of regions of endochondral ossification. The clinical aggressiveness of Clival GCT is strongly related to its complicated anatomical location, which implies multiple cranial nerve involvement with subsequent headache, decreased vision, visual field defect, diplopia, ophthalmoplegia, and deafness [18, 20]. The traditional approaches for diagnosis of GCT of the clivus comprise skull X-ray, computed tomography and magnetic resonance imaging but imaging studies alone are not enough to distinguish Clival GCT from other bone lesions. Indeed, the definitive diagnosis is only postoperative, based on biopsy findings and the main histomorphologic feature is the presence of giant cells similar to osteoclasts [20]. However, multinucleated osteoclast-like giant cells can be found in several malignant and nonmalignant bone lesions (i.e. osteosarcomas, chondroblastomas, giant cell granulomas, giant cell reparative granulomas, and brown tumours of hyperparathyroidism); therefore, the postoperative histopathology can be ambiguous and Clival GCT can be easily misdiagnosed [16, 20, 24].
In this study, we unveiled the genetic basis of the giant cell tumour of the clivus, analysing the tumour specimens of two patients affected by this aggressive bone lesion. We analysed the coding regions of H3F3A, gene mutated in most cases of giant cell tumour of long bones. Indeed, since 2013, H3.3 mutations have been identified in more than 90% of all GCT cases analysed, with the p.Gly34Trp as the most frequently harboured mutation [4, 5, 70,71,72]. We also examined the mutational status of H3F3B, IDH1 and IDH2 that, though more rarely, have been associated to GCT [3, 5, 28]. Besides, we investigated the presence of mutation in the coding region of the ZNF687 gene that we recently identified in giant cell tumour complicating Paget’s disease of bone [6]. The latter is a localised disorder of excessive and abnormal bone remodelling that can also affect the skull base, especially the clivus and surrounding sphenoid bone structures [73]. Considering this frequent localisation (42%) as well as the evidence that the tumour biopsy of patient 1 was composed of giant cells whose size resembled that found in pagetic GCT, we sought to investigate whether these two clival masses could represent GCT degeneration of pagetic lesions [7]. Interestingly, we highlighted that both tumour samples harbour the p.Gly34Trp mutation in the H3F3A gene and demonstrated that the change was somatically acquired, as it was not found in the peripheral blood. Moreover, our immunofluorescence analysis further confirmed the restriction of the H3F3A mutation to the stromal cell population that was abundant in both tumour biopsies. Consequently, even though in a relatively low number of samples, we demonstrated that two cases of GCT affecting the clivus are genetically defined by mutation in the H3F3A gene and hence, Clival GCT can be considered a typical giant cell tumour with a peculiar anatomical localisation. Therefore, the mutational analysis of the H3F3A gene is an accurate tool for the diagnosis of Clival GCT that can be combined with the histological analysis of the tumour sample in order to distinguish the giant cell tumour from other osteoclast-rich tumours. In agreement, it has been recently demonstrated that H3F3A mutations were not detectable in neither giant cell-rich sarcomas nor giant cell-rich benign lesions, whereas almost all GCT tissues carried H3F3A mutation [4, 71]. While the H3F3A genetic screening allows to distinguish GCT from other giant cell-rich lesions, the histology-based diagnostic algorithm that we generated in this study may be useful to differentiate other morphologically overlapping lesions arising within the clivus.
We also found a strong Tenascin-C immunoreactivity in the two Clival GCT biopsies, indicative of an aggressive tumour with a tendency to recur in situ, as this extracellular glycoprotein is highly expressed in the microenvironment of most solid tumours to promote migration and epithelial-mesenchymal transition [74]. As a matter of fact, the patient 1 have had a local recurrence of the tumour and has been recently planned for resurgery. However, Tenascin-C immunoreactivity can also be found in central giant cell granulomas, making it not a suitable histological marker to distinguish GCTs from other giant cell-rich lesions [75]. Nevertheless, the expression of Tenascin-C in both tumour samples further links the Clival GCT to the GCT of long bones, where its intense expression in the extracellular matrix has been clearly described by us and others [7, 74, 75]. Finally, the ability of mononuclear cells within Clival GCT to produce high levels of RANKL, mimicking what happens in GCT of long bones, led us to speculate that Denosumab treatment could represent an effective therapy for Clival GCT.
Overall, these results demonstrate that Clival GCTs are caused by the same genetic defect of GCTs of long bones and allowed us to hypothesize that the two tumour types can be managed using the same approaches. The gold standard for management of this tumour contemplates the surgical resection, followed by adjuvant treatment with chemotherapy or radiotherapy [15,16,17, 20, 24]. However, no effective chemotherapeutic agents have yet been identified and some patients developed osteosarcoma transformation at sites of previous irradiation [15, 23, 76]. To date, only one case of Clival GCT has been treated with Denosumab after surgical resection by endoscopic endonasal transsphenoidal surgery and the treatment proved to be effective in preventing tumour growth [23].
Conclusions
We conclude that the Clival GCT is genetically defined by somatic mutation in the H3F3A gene. Our data also suggest that the GCT of the clivus can be treated on the same principles as that of GCT of long bones and that the relatively more aggressive phenotype of Clival GCTs should only be addressed to their anatomical location.
Abbreviations
- CT:
-
Computed tomography
- FFPE:
-
Formalin Fixed Paraffin Embedded
- GCT:
-
Giant cell tumour
- H3F3A :
-
Human gene encoding H3 Histone Family Member 3A
- H3F3B :
-
Human gene encoding H3 Histone Family Member 3B
- IDH1 :
-
Human gene encoding Isocitrate dehydrogenase 1
- IDH2 :
-
Human gene encoding Isocitrate dehydrogenase 2
- MRI:
-
Magnetic resonance imaging
- RANK:
-
Receptor Activator of Nuclear Factor κ B
- RANKL:
-
Receptor activator of nuclear factor kappa-B ligand
- TRAP:
-
Tartrate-resistant acid phosphatase
- ZNF687 :
-
Human gene encoding zinc finger protein 687
References
Campanacci M, Baldini N, Boriani S, Sudanese A. Giant-cell tumour of bone. J Bone Joint Surg Am. 1987;69:106–14.
Morgan T, Atkins GJ, Trivett MK, Johnson SA, Kansara M, Schlicht SL, et al. Molecular profiling of giant cell tumour of bone and the osteoclastic localization of ligand for receptor activator of nuclear factor kappaB. Am J Pathol. 2005;167:117–28.
Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumour of bone. Nat Genet. 2013;45:1479–82.
Presneau N, Baumhoer D, Behjati S, Pillay N, Tarpey P, Campbell PJ, et al. Diagnostic value of H3F3A mutations in giant cell tumour of bone compared to osteoclast-rich mimics. J Pathol Clin Res. 2015;1:113–23.
Cleven AH, Höcker S, Briaire-de Bruijn I, Szuhai K, Cleton-Jansen AM, Bovée JV. Mutation analysis of H3F3A and H3F3B as a diagnostic tool for Giant cell tumour of bone and Chondroblastoma. Am J Surg Pathol. 2015;39:1576–83.
Divisato G, Formicola D, Esposito T, Merlotti D, Pazzaglia L, Del Fattore A, et al. ZNF687 mutations in severe Paget disease of bone associated with Giant cell tumour. Am J Hum Genet. 2016;98:275–86.
Divisato G, Scotto di Carlo F, Pazzaglia L, Rizzo R, Coviello DA, Benassi MS, et al. The distinct clinical features of Giant cell tumour of bone in pagetic and non-pagetic patients are associated with genetic, biochemical and histological differences. Oncotarget. 2017; https://doi.org/10.18632/oncotarget.18670.
Gouin F, Dumaine V. French Sarcoma and Bone tumor study groups GSF-GETO. Local recurrence after curettage treatment of giant cell tumors in peripheral bones: retrospective study by the GSF-GETO (French sarcoma and bone tumor study groups). Orthop Traumatol Surg Res. 2013;99:S313–8.
Gaston CL, Grimer RJ, Parry M, Stacchiotti S, Dei Tos AP, Gelderblom H, et al. Current status and unanswered questions on the use of Denosumab in giant cell tumor of bone. Clin Sarcoma Res. 2016;6:15.
Thomas D, Henshaw R, Skubitz K, Chawla S, Staddon A, Blay JY, et al. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010;11:275–80.
Wülling M, Engels C, Jesse N, Werner M, Delling G, Kaiser E. The nature of giant cell tumour of bone. J Cancer Res Clin Oncol. 2001;127:467–74.
Goldenberg RR, Campbell CJ, Bonfiglio M. Giant-cell tumour of bone. An analysis of two hundred and eighteen cases. J Bone Joint Surg Am 1970;52:619–664.
Larsson SE, Lorentzon R, Boquist L. Giant-cell tumour of bone. A demographic, clinical, and histopathological study of all cases recorded in the Swedish Cancer registry for the years 1958 through 1968. J Bone Joint Surg Am. 1975;57:167–73.
Bertoni F, Unni KK, Beabout JW, Ebersold MJ. Giant cell tumour of the skull. Cancer. 1992;70:1124–32.
Sasagawa Y, Tachibana O, Shiraga S, Takata H, Kinoshita E, Nojima T, et al. Secondary malignant giant cell tumour of the clivus: case report. Clin Neurol Neurosurg. 2012;114:786–8.
Mahale A, K V N D, Pai M, Poornima V, Sahu KKMRI. Sequence and characteristic features in 'giant cell tumour' of clivus. J Clin Diagn Res. 2013;7:1197–200.
Roy S, Joshi NP, Sigamani E, Malik A, Sharma MC, Mohanti BK, et al. Clival giant cell tumour presenting with isolated trigeminal nerve involvement. Eur Arch Otorhinolaryngol. 2013;270:1167–71.
Iacoangeli M, Di Rienzo A, Re M, Alvaro L, Nocchi N, Gladi M, et al. Endoscopic endonasal approach for the treatment of a large clival giant cell tumour complicated by an intraoperative internal carotid artery rupture. Cancer Manag Res. 2013;5:21–4.
Agrawal A, Gali R, Shanthi V, Ramakrishna BA, Mohan KV. Giant cell tumour of the clivus with presence of epithelioid histiocytes. Asian J Neurosurg. 2014;9:48–9.
Zhao J, Qian T, Zhi Z, Li Q, Kang L, Wang J, et al. Giant cell tumour of the clivus: a case report and review of the literature. Oncol Lett. 2014;8:2782–6.
Le J, Chaiyasate K, Donev K, Fahim DK. A rare case of giant cell tumour involving the clivus resected through Le fort I osteotomy and median maxillotomy. Surg Neurol Int. 2015;6:26.
Shibao S, Toda M, Yoshida K. Giant cell tumours of the clivus: case report and literature review. Surg Neurol Int. 2015;6:S623–7.
Inoue A, Ohnishi T, Kohno S, Nishikawa M, Nishida N, Ohue S. Role of Denosumab in endoscopic Endonasal treatment for juvenile Clival Giant cell tumour: a case report and review of the literature. World Neurosurg. 2016;91:674.e1–6.
Patibandla MR, Thotakura AK, Rao MN, Addagada GC, Nukavarapu MC, Panigrahi MK, et al. Clival giant cell tumour - a rare case report and review of literature with respect to current line of management. Asian J Neurosurg. 2017;12:78–81.
Zorlu F, Selek U, Soylemezoglu F, Oge K. Malignant giant cell tumor of the skull base originating from clivus and sphenoid bone. J Neuro-Oncol. 2006;76:149–52.
Gupta R, Mohindra S, Mahore A, Mathuriya SN, Radotra BD. Giant cell tumour of the clivus. Br J Neurosurg. 2008;22:447–9.
Lüke J, von Baer A, Schreiber J, Lübbehüsen C, Breining T, Mellert K, et al. H3F3A mutation in giant cell tumour of the bone is detected by immunohistochemistry using a monoclonal antibody against the G34W mutated site of the histone H3.3 variant. Histopathology. 2017;71:125–33.
Kato Kaneko M, Liu X, Oki H, Ogasawara S, Nakamura T, Saidoh N, et al. Isocitrate dehydrogenase mutation is frequently observed in giant cell tumour of bone. Cancer Sci. 2014;105:744–8.
Dahlin DC. Caldwell lecture. Giant cell tumor of bone: highlights of 407 cases. AJR Am J Roentgenol. 1985;144:955–60.
Picci P, Manfrini M, Fabbri N, Gambarotti M, Vanel D. Atlas of Musculoskeletal Tumors and Tumorlike Lesions. Switzerland: Springer International Publishing; 2014.
Potter GD, McClennan BL. Malignant giant cell tumor of the sphenoid bone and its differential diagnosis. Cancer. 1970;25:167–70.
Doshi R, Chaudhari AB, Thomson G. Giant cell tumor of the sphenoid bone. Can J Neurol Sci. 1977;4:213–6.
Wilbur AC, Choi KH, Tan WS, Jafar JJ, Spigos DG. Giant cell tumor of the sphenoid bone mimicking a pituitary tumor. AJNR Am J Neuroradiol. 1986;7:361–2.
Weber AL, Hug EB, Muenter MW, Curtin HD. Giant-cell tumors of the sphenoid bone in four children: radiological, clinical, and pathological findings. Skull Base Surg. 1997;7:163–73.
Kattner KA, Stroink A, Gupta K, Fukushima T, Li C. Giant cell tumor of the sphenoid bone. Skull Base Surg. 1998;8:93–7.
Krishnan G, Narendra K, Sundhar K. Aggressive Osteoclastoma of sphenoid sinus: a rare surgical case report. Otorhinolaryngology clinics - An International Journal. 2016;8:68–71.
Goto Y, Furuno Y, Kawabe T, Ohwada K, Tatsuzawa K, Sasajima H, et al. Treatment of a skull-base giant cell tumor with endoscopic endonasal resection and denosumab: case report. J Neurosurg. 2017;126:431–4.
Motomochi M, Handa Y, Makita Y, Hashi K. Giant cell tumor of the skull. Surg Neurol. 1985;23:25–30.
Büter JJ, Chilla R. Giant cell tumor of the temporal bone (osteoclastoma). Eur Arch Otorhinolaryngol. 1997;254:298–300.
Tsai Y-F, Liang-Kong C, Su C-T, Lee C-C, Wai C-P, Chen S-Y. Giant cell tumor of the Skull Base: a case report. Chin J Radiol. 2000;25:223–7.
Venkatesh MD, Vijaya N, Girish N, Galagali JR. Giant cell tumor of temporal bone: a case report. Med J Armed Forces India. 2012;68:392–4.
Hsu S-W, Hueng D-Y, Lin H-C, Liu M-Y, Ma H-I, Hsia C-C. Giant cell tumor of the temporal bone. Formosan Journal of Surgery. 2013;46:30–2.
Billingsley JT, Wiet RM, Petruzzelli GJ, Byrne R. A locally invasive giant cell tumor of the skull base: case report. J Neurol Surg Rep. 2014;75:e175–9.
Byun JH, Park KB, Ko JS, Ahn SK. Giant cell tumor of infratemporal Fossa and mandibular condyle: a case report. J Int Adv Otol. 2015;11:88–91.
Tamura R, Miwa T, Shimizu K, Mizutani K, Tomita H, Yamane N, et al. Giant cell tumor of the skull: review of the literature. J Neurol Surg A Cent Eur Neurosurg. 2016;77:239–46.
Sang WH, Ha Young C. Malignant Giant cell tumor of the skull. J Korean Neurosurg Soc. 2004;36:324–7.
Lu ZH, Yao ZW. Giant cell tumour of the posterior cranial fossa: a case report. Br J Radiol. 2011;84:e206–9.
Modkovski R, Elliott R, Rubin B, Zagzag D, Jafar J, Mikolaenko I. Giant cell tumor of the occipital bone and secondary aneurysmal bone cyst: case report and review of literature. The internet. J Neurosurg. 2009;7:1–7.
Uslu GH, Canyilmaz E, Yöney A, Aydin S, Sahbaz A, Sari A. Giant cell tumor of the occipital bone: a case report and review of the literature. Oncol Lett. 2014;8:151–4.
Tang PH, Mettu P, Maltry AC, Harrison AR, Mokhtarzadeh A. Giant cell tumor of the frontal bone presenting as an orbital mass. Ophthalmol Ther. 2017;6:215–20.
Alzarei A, Assiri M, R NB, Aljoraebi W, Sumaily I. Giant cell tumor of the frontal sinus: case report. International Journal of Otorhinolaryngology and Head and Neck Surgery. 2017;3:725–7.
Erdem E, Angtuaco EC, Van Hemert R, Park JS, Al-Mefty O. Comprehensive review of intracranial chordoma. Radiographics. 2003;23:995–1009.
Chambers KJ, Lin DT, Meier J, Remenschneider A, Herr M, Gray ST. Incidence and survival patterns of cranial chordoma in the United States. Laryngoscope. 2014;124:1097–102.
Korten AG, ter Berg HJ, Spincemaille GH, van der Laan RT, Van de Wel AM. Intracranial chondrosarcoma: review of the literature and report of 15 cases. J Neurol Neurosurg Psychiatry. 1998;65:88–92.
Brackmann DE, Teufert KB. Chondrosarcoma of the skull base: long-term follow-up. Otol Neurotol. 2006;27:981–91.
Almefty K, Pravdenkova S, Colli BO, Al-Mefty O, Gokden M. Chordoma and chondrosarcoma: similar, but quite different, skull base tumors. Cancer. 2007;110:2457–67.
Rosenberg AE, Nielsen GP, Keel SB, Renard LG, Fitzek MM, Munzenrider JE, et al. Chondrosarcoma of the base of the skull: a clinicopathologic study of 200 cases with emphasis on its distinction from chordoma. Am J Surg Pathol. 1999;23:1370–8.
NG HK, Wong AT. Expression of epithelial and extracellular matrix protein markers in meningiomas. Histopathology. 1993;22:113–25.
Kawase T, Shiobara R, Ohira T, Toya S. Developmental patterns and characteristic symptoms of petroclival meningiomas. Neurol Med Chir (Tokyo). 1996;36:1–6.
Liu Y, Sturgis CD, Bunker M, Saad RS, Tung M, Raab SS, Silverman JF. Expression of cytokeratin by malignant meningiomas: diagnostic pitfall of cytokeratin to separate malignant meningiomas from metastatic carcinoma. Mod Pathol. 2004;17:1129–33.
Guo Z, Hu K, Zhao B, Bian E, Ni S, Wan J. Osteosarcoma of the skull base: an analysis of 19 cases and literature review. J Clin Neurosci. 2017;44:133–42.
Yoshida H, Adachi H, Hamada Y, Aki T, Yumoto T, Morimoto K, et al. Osteosarcoma. Ultrastructural and immunohistochemical studies on alkaline phosphatase-positive tumor cells constituting a variety of histologic types. Acta Pathol Jpn. 1988;38:325–38.
Karras CL, Abecassis IJ, Abecassis ZA, Adel JG, Bit-Ivan EN, Chandra RK, et al. Clival ectopic pituitary adenoma mimicking a Chordoma: case report and review of the literature. Case Rep Neurol Med. 2016;2016:8371697.
Grau S, Schueller U, Weiss C, Tonn JC. Primary meningeal T-cell lymphoma at the clivus mimicking a meningioma. World Neurosurg. 2010;74:513–6.
Kalwani N, Remenschneider AK, Faquin W, Ferry J, Holbrook EH. Plasmacytoma of the Clivus presenting as bilateral sixth nerve palsy. J Neurol Surg Rep. 2015;76:e156–9.
Boyd SD, Natkunam Y, Allen JR, Warnke RA. Selective immunophenotyping for diagnosis of B-cell neoplasms: immunohistochemistry and flow cytometry strategies and results. Appl Immunohistochem Mol Morphol. 2013;21:116–31.
Pallini R, Sabatino G, Doglietto F, Lauretti L, Fernandez E, Maira G. Clivus metastases: report of seven patients and literature review. Acta Neurochir. 2009;151:291–6.
Deconde AS, Sanaiha Y, Suh JD, Bhuta S, Bergsneider M, Wang MB. Metastatic disease to the clivus mimicking clival chordomas. J Neurol Surg B Skull Base. 2013;74:292–9.
Yu WYH, Tierney LM. HCC without cirrhosis metastasizing to the clivus. Int Canc Conf J. 2015;4:181.
Amary F, Berisha F, Ye H, Gupta M, Gutteridge A, Baumhoer D, et al. H3F3A (histone 3.3) G34W immunohistochemistry: a reliable marker defining benign and malignant Giant cell tumor of bone. Am J Surg Pathol. 2017;41:1059–68.
Righi A, Mancini I, Gambarotti M, Picci P, Gamberi G, Marraccini C, et al. Histone 3.3 mutations in giant cell tumor and giant cell-rich sarcomas of bone. Hum Pathol. 2017;68:128–35.
Kervarrec T, Collin C, Larousserie F, Bouvier C, Aubert S, Gomez-Brouchet A, et al. H3F3 mutation status of giant cell tumors of the bone, chondroblastomas and their mimics: a combined high resolution melting and pyrosequencing approach. Mod Pathol. 2017;30:393–406.
Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int. 2012;91:97–113.
Pazzaglia L, Conti A, Chiechi A, Novello C, Magagnoli G, Astolfi A, et al. Differential gene expression in classic giant cell tumours of bone: tenascin C as biological risk factor for local relapses and metastases. Histopathology. 2010;57:59–72.
Aragão Mdo S, Piva MR, Nonaka CF, Freitas Rde A, de Souza LB, Pinto LP. Central giant cell granuloma of the jaws and giant cell tumor of long bones: an immunohistochemical comparative study. J Appl Oral Sci. 2007;15:310–6.
Bertoni F, Bacchini P, Staals EL. Malignancy in giant cell tumor of bone. Cancer. 2003;97:2520–9.
Gray H, Carter HV. Anatomy of the human body. 20th ed. Lea & Febiger; 1918
Acknowledgements
We thank Dr. Ennio Scotto di Carlo for helpful suggestions. We gratefully thank the patients for their participation to our study.
Funding
This work was supported by the Italian Association for Cancer Research (AIRC), Grant IG-2014 n.15707 (F.G.).
Availability of data and materials
Data and materials are available from the authors upon reasonable request.
Author information
Authors and Affiliations
Contributions
Study design: FG. Study conduct: FSdC, GD, MI. Data collection: FSdC, GD, TE, FG. Data analysis: FSdC, GD, TE, FG. Data interpretation: FG. Drafting manuscript: FSdC, GD, FG. Approving final version of manuscript: FSdC, GD, MI, TE, FG. FG takes responsibility for the integrity of the data analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Authors’ information
FSdC and GD are PhD student and Postdoc in Gianfrancesco’s group working on bone disorders and their neoplastic degenerations. MI is a Medical Doctor at Department of Neurosurgery, Umberto I General Hospital, Università Politecnica delle Marche, Ancona, Italy. TE and FG are full researchers of National Research Council of Italy at Institute of Genetics and Biophysics “Adriano Buzzati-Traverso” of Naples. Gianfrancesco’s research activity is focused on the identification of causative genes for human diseases.
Ethics approval and consent to participate
This study was approved by Ethics Committee for Biomedical Activities “Carlo Romano”, International Office for Bioethics Research Federico II University of Naples. Written informed consent was obtained individually from both patients.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Scotto di Carlo, F., Divisato, G., Iacoangeli, M. et al. The identification of H3F3A mutation in giant cell tumour of the clivus and the histological diagnostic algorithm of other clival lesions permit the differential diagnosis in this location. BMC Cancer 18, 358 (2018). https://doi.org/10.1186/s12885-018-4291-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-018-4291-z