
Abstract
Background
Lung cancer patients with an activating mutation in the EGFR (epidermal growth factor receptor) can develop resistance to erlotinib treatment, which is often mediated by the T790M resistance mutation in EGFR. The difficulties in obtaining biopsies at progression make it challenging to investigate the appearance of the T790M mutation at progression in large patient cohorts. We have used cell free DNA (cfDNA) from patients treated with erlotinib to investigate if the development of a T790M mutation coincides with the presence of an activating EGFR mutation in the pre-treatment blood sample.
Methods
A cohort of 227 NSCLC (non-small cell lung cancer) adenocarcinoma patients was treated with erlotinib irrespective of EGFR-mutational status. Blood samples were drawn immediately before erlotinib treatment was initiated and again at progression. The cobas® EGFR Mutation Test v2 designed for cfDNA was used to identify 42 EGFR mutations.
Results
Of the 227 NSCLC patients, blood samples were available from 144 patients both before erlotinib treatment and at progression (within 1 month before or after clinical progression). One hundred and twenty-eight of the 144 were wild-type EGFR before treatment, and we demonstrate that the T790M mutation was not present at progression in any of these. In contrast, in the 16 patients with an activating EGFR mutation in the pre-treatment blood sample six patients (38%) were identified with a T790M mutation in the progression blood sample.
Conclusion
The T790M resistance mutation is only found in the cfDNA of erlotinib-treated NSCLC patients if they have an activating EGFR mutation before treatment.
Similar content being viewed by others


Background
A subgroup of NSCLC patients have activating mutations in the EGFR gene, primarily in exon 19 and 21 [1,2,3]. This group responds well to EGFR-directed tyrosine kinase inhibitors (TKIs), like erlotinib and gefitinib, and today these agents are part of standard care. Despite an initial response, all patients eventually acquire resistance to the treatment [4,5,6]. One defined mechanism causing resistance to TKI treatment is the development of the T790M mutation in exon 20 of the EGFR gene [7, 8]. Additionally, it has been shown that amplification of the MET (met proto-oncogene (hepatocyte growth factor receptor)) gene or increased expression of HGF (hepatocyte growth factor) can cause resistance (reviewed by Tartarone et al. [9]).
Apart from the NSCLC patients with activating EGFR mutations, it has been suggested that a subgroup of the EGFR wild-type patients also experience benefit from erlotinib treatment [10]. These patients ultimately develop resistance as well. Not much is known about the resistance mechanisms for this patient group. Generally, investigation of resistance mechanisms for both EGFR wild type and mutation-positive patients is complicated, since re-biopsies from patients with progression are not taken systematically.
In the present study, we have taken advantage of a new possibility of detecting the T790M resistance mutation in a blood sample [11]. We used the cobas® EGFR Mutation Test v2 designed for cell free DNA (cfDNA) to investigate the presence of the T790M mutation in the cfDNA from NSCLC patients treated with erlotinib.
Methods
Patients and blood samples
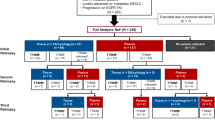
From a previously collected cohort, 227 NSCLC adenocarcinoma patients treated with erlotinib from October 2008 to December 2012 were selected based on the presence of both a biopsy and paired blood samples isolated during treatment [10]. The project was approved by the Central Denmark Region Commitees on Biomedical Research Ethics (M-20080012). All included patients gave written consent.
Blood samples were collected immediately before erlotinib treatment was initiated, and again when treatment was terminated due to progression as defined by RECIST (response evaluation criteria in solid tumors) version 1.1 criteria or by clinical deterioration defined by the treating physician. Patients without progression at first follow-up with CT-scan (3 months after therapy initiation) were considered responders [10].
Of these 227 patients, 144 patients had paired blood samples before erlotinib treatment and within 1 month before or after progression. A total of 16 of the 144 patients had an activating mutation in the EGFR gene in the pre-treatment plasma DNA sample, as well as in the tumor biopsy taken at diagnosis, whereas 128 were EGFR wild type. None of the patients had a T790 M mutation in their tumor before treatment. Patient characteristics can be seen in Table 1. Blood and biopsy sampling, and identification of EGFR mutations using the cobas® EGFR Mutation Test v2 (Roche Molecular Systems, Inc., CA, USA) have previously been described [11, 12]. The test contains positive and negative controls and all runs passed this quality check. CfDNA was isolated from 2 ml of plasma and eluted in 100 μL with the cobas DNA sample preparation kit (Roche).
Droplet digital PCR
Droplet digital PCR was performed on the QX200 droplet digital PCR system (BioRad). Five μL of the cfDNA isolated as described above was used for ddPCR and all samples were analyzed in triplicate. Validated kits for both the wild type and the T790M-mutated EGFR were purchased from BioRad (cat #10031250 and cat #10031247) and the assay was performed with the ddPCR Supermix For Probes Reagents (BioRad, cat #186–3025). Results were analyzed with the QuantaSoft software (Bio-Rad).
Cell lines and reagents
Wild-type EGFR adenocarcinoma NSCLC cell lines Calu-3 (ATCC® HTB55™), H358 (ATCC® CRL-5807™), H1568 (ATCC® CRL-5876™), and H1666 (ATCC® CRL-5885™) were purchased at ATCC. Calu-3 was cultured in MEM media (Gibco, Lifetechnologies) and H358, H1568, and H1666 were cultured in RPMI-1640 media (Gibco, Lifetechnologies) according to the manufacturer’s instructions. Erlotinib-resistant cell lines were established by two methods; step-wise escalation of drug (ER), where the drug concentration was escalated from 10 nM to 5 μM, and high dose drug (HD), where the drug concentration was fixed at 5 μM. Cells were considered resistant after 4–6 months when they were able to grow in 5 μM erlotinib (Selleckchem). Cell viability studies were performed using CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega) following manufacturer’s recommendations.
Statistics
The Fisher’s exact test was used to assess the hypothesis of no difference in risk of gaining the T790M mutation between patients with and without an activating EGFR mutation. All statistics were calculated in Stata 13. Data obtained from cell viability studies are presented in graphs produced in Prism 6.
Results
One hundred and twenty-eight patients with wild-type EGFR in the pre-treatment blood sample were identified among the 144 included patients. None of these patients had developed the T790M resistance mutation in the blood sample taken at progression (Table 2). Among the 16 patients harboring an activating EGFR mutation in the pre-treatment blood sample, the T790M mutation was found in 6 (38%) of the blood samples taken at the time of progression. Table 2 shows the frequency of the T790M mutation in blood samples taken at the time of progression in patients with or without activating EGFR mutations in the pre-treatment blood sample. Statistical analysis demonstrated that the T790M mutation was preferentially associated with the presence of an activating EGFR mutation (p < 0.0001, Fisher’s exact test). Also, we investigated the distribution of the T790M mutation in patients with defined clinical response to treatment (see Table 3). Again, the T790M mutation was associated with an activating mutation in EGFR (p < 0.0001, Fisher’s exact test).
To further substantiate these results, we performed droplet digital PCR (ddPCR) with DNA from the paired blood samples of the 16 patients with activating EGFR mutations to detect the T790M mutation. The T790M mutation was not present in any of the pre-treatment blood samples as seen with the Cobas assay. Complete concordance was also apparent between the results from the cobas® EGFR Mutation Test v2 and ddPCR in the progression blood samples (Table 4).
To investigate whether EGFR wild-type NSCLC cell lines developed T790M-mediated resistance, we established two different in vitro cell line models of erlotinib resistance with four EGFR wild-type NSCLC cell lines. Erlotinib response of all parental and resistant cell lines can be seen in Additional file 1: Figure S1. None of the eight resulting erlotinib-resistant cell lines developed the T790M mutation, or any other secondary EGFR mutation, as a resistance mechanism to erlotinib when analyzed with the cobas® EGFR Mutation Test v2.
Our results demonstrate that the T790M mutation is only detected in the progression blood samples of patients harboring a pre-treatment activating EGFR mutation and not in either patients or cell lines with wild-type EGFR.
Discussion
Emergence of T790M-driven resistance during EGFR-directed TKI treatment in patients with activating EGFR mutations is well described. The presence of T790M in EGFR wild-type patients treated with TKI has not been investigated in detail. Traditionally, re-biopsies taken at the time of progression would be needed to study this. However, the opportunity to study tumor DNA in blood samples offers a possibility to overcome this limitation. Here we present data showing that the T790M mutation is not found in the blood of 128 EGFR wild-type patients at the time of progression. In comparison, the mutation is present in 38% (6/16) of patients with an activating EGFR mutation before treatment.
We further supported our findings by in vitro studies, where we generated EGFR wild-type erlotinib-resistant cell lines, none of which acquired the T790M mutation as a resistance mechanism to erlotinib. In contrast, the development of T790M mutations in lung cancer cell lines harboring an activating EGFR mutation is well described [13,14,15,16].
To our best knowledge, this is the first study to investigate the presence of EGFR T790M as resistance mechanism in a wild-type EGFR patient cohort. Prior studies have preferably investigated epithelial-to-mesenchymal transition (EMT) as a mechanism of both acquired and intrinsic resistance mechanism. The majority of these find that EGFR TKI sensitivity both in vitro and in vivo is correlated with an epithelial phenotype [17,18,19,20], though conflicting results has also been presented [21]. Furthermore, an in vitro study have shown that activation of insulin-like growth factor 1 receptor may act as mechanism of acquired resistance to EGFR TKI in a wild-type EGFR NSCLC cell line [22]. In the present study, we have not investigated these mechanisms but focused on the observation that the T790 M mutation in EGFR does not appear in an EGFR wild-type background.
Conclusion
Our results suggest that erlotinib-treated patients with wild-type EGFR develop resistance through other mechanisms than T790M, and molecular diagnostics aimed at identifying the responsible events should not focus on T790M.
Abbreviations
- cfDNA:
-
cell free DNA
- ddPCR:
-
droplet digital PCR
- EGFR:
-
Epidermal growth factor receptor
- HGF:
-
Hepatocyte growth factor
- MET:
-
Met proto-oncogene (hepatocyte growth factor receptor)
- NSCLC:
-
Non-small cell lung cancer
- RECIST:
-
Response evaluation criteria in solid tumors
References
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39.
Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500.
Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11.
Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Jänne PA, Lynch T, Johnson BE, Miller VA. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28:357–60.
Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinohita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, Ando M, Miyazawa H, Tanaka T, Saijo Y, Hagiwara K, Morita S, Nukiwa T. Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–8.
Mok TS, Wu Y-L, Thongprasert S, Yang C-H, Chu D-T, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang J-J, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57.
Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73.
Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92.
Tartarone A, Lazzari C, Lerose R, Conteduca V, Improta G, Zupa A, Bulotta A, Aieta M, Gregorc V. Mechanisms of resistance to EGFR tyrosine kinase inhibitors gefitinib/erlotinib and to ALK inhibitor crizotinib. Lung Cancer. 2013;81:328–36.
Weber B, Hager H, Sorensen BS, McCulloch T, Mellemgaard A, Khalil AA, Nexo E, Meldgaard P. EGFR mutation frequency and effectiveness of erlotinib: a prospective observational study in Danish patients with non-small cell lung cancer. Lung Cancer. 2014;83:224–30.
Weber B, Meldgaard P, Hager H, Wu L, Wei W, Tsai J, Khalil A, Nexo E, Sorensen BS. Detection of EGFR mutations in plasma and biopsies from non-small cell lung cancer patients by allele-specific PCR assays. BMC Cancer. 2014;14:294.
Sorensen BS, Wu L, Wei W, Tsai J, Weber B, Nexo E, Meldgaard P. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor-sensitizing and resistance mutations in the plasma DNA of patients with advanced non-small cell lung cancer during treatment with erlotinib. Cancer. 2014;120:3896–901.
Ogino A, Kitao H, Hirano S, Uchida A, Ishiai M, Kozuki T, Takigawa N, Takata M, Kiura K, Tanimoto M. Emergence of epidermal growth factor receptor T790M mutation during chronic exposure to gefitinib in a non small cell lung cancer cell line. Cancer Res. 2007;67:7807–14.
Ware KE, Hinz TK, Kleczko E, Singleton KR, L a M, B a H, Cummings CT, Graham DK, Astling D, Tan a-C, Heasley LE. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogene. 2013;2:e39.
Shien K, Toyooka S, Yamamoto H, Soh J, Jida M, Thu KL, Hashida S, Maki Y, Ichihara E, Asano H, Tsukuda K, Takigawa N, Kiura K, Gazdar AF, Lam WL, Miyoshi S. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 2013;73:3051–61.
Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin Y-L, Pan Y, Wang L, de Stanchina E, Shien K, Aoe K, Toyooka S, Kiura K, Fernandez-Cuesta L, Fidias P, Yang JC-H, Miller VA, Riely GJ, Kris MG, Engelman JA, Vnencak-Jones CL, Dias-Santagata D, Ladanyi M, Pao W. PNAS Plus: lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci. 2012;109:E2127–33.
Ren S, Su C, Wang Z, Li J, Fan L, Li B, Li X, Zhao C, Wu C, Hou L, He Y, Gao G, Chen X, Ren J, Li A, Xu G, Zhou X, Zhou C, Schmid-Bindert G. Epithelial phenotype as a predictive marker for response to EGFR-TKIs in non-small cell lung cancer patients with wild-type EGFR. Int J Cancer. 2014;135:2962–71.
Frederick BA, Helfrich BA, Coldren CD, Zheng D, Chan D, Bunn PA, Raben D. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther. 2007;6:1683–91.
Chen B, Xiao F, Li B, Xie B, Zhou J, Zheng J, Zhang W. The role of epithelial-mesenchymal transition and IGF-1R expression in prediction of gefitinib activity as the second-line treatment for advanced nonsmall-cell lung cancer. Cancer Investig. 2013;31:454–60.
Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, Nilsson MB, Gudikote J, Tran H, Cardnell RJG, Bearss DJ, Warner SL, Foulks JM, Kanner SB, Gandhi V, Krett N, Rosen ST, Kim ES, Herbst RS, Blumenschein GR, Lee JJ, Lippman SM, Ang KK, Mills GB, Hong WK, Weinstein JN, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–90.
Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, Modrusan Z, Lin C-Y, O’Neill V, Amler LC. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11(24 Pt 1):8686–98.
Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, Yatabe Y, Sekido Y, Mitsudomi T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011;6:1152–61.
Acknowledgements
The authors wish to acknowledge skillful technical assistance from laboratory technicians Birgit Westh Mortensen and Lene Dabelstein Petersen.
Funding
None.
Availability of data and materials
The datasets analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
CD performed the experiments and drafted the manuscript. ATM performed the experiments and helped to draft the manuscript. BW participated in the design of the study and collected patient data. LW participated in the design of the study and helped to draft the manuscript. PM was involved in the design of the study, the statistics and helped in drafting the manuscript. BS was involved in the design of the study, the statistics and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All patients gave informed written consent and the project was in compliance with the Helsinki Declaration. The Central Denmark Region Committee on Biomedical Research Ethics approved the project (M-20080012).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1
Figure S1. The erlotinib response of parental and resistant cell lines. A and B: H1568 parental cell line along with H1568 ER and HD. C and D: H1666 parental cell line and H1666 ER and HD. E and F: Calu-3 parental cell line and Calu-3 ER and HD. G and H: H358 parental cell line and H358 ER and HD. ER: erlotinib-resistant cell lines generated by the stepwise escalation method. HD: erlotinib-resistant cell lines generated by the high dose method. (TIFF 1290 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Demuth, C., Madsen, A.T., Weber, B. et al. The T790M resistance mutation in EGFR is only found in cfDNA from erlotinib-treated NSCLC patients that harbored an activating EGFR mutation before treatment. BMC Cancer 18, 191 (2018). https://doi.org/10.1186/s12885-018-4108-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-018-4108-0