
Abstract
Background
Real-time polymerase chain reaction (PCR) has become an increasingly important technique for gene expression profiling because it can provide insights into complex biological and pathological processes and be used to predict disease or treatment outcomes. Although normalized data are necessary for an accurate estimation of mRNA expression levels, several pieces of evidence suggest that the expression of so-called housekeeping genes is not stable. This study aimed to validate reference genes for the normalization of real-time PCR in an N-methyl-N-nitrosourea (MNU)-induced T-cell lymphoma mouse model.
Methods
T-cell lymphomas were generated in p53-deficient mice by treatment with 37.5 mg/kg MNU. Thymus and spleen were identified as the primary target organs with the highest incidences of lymphomas. We analyzed the RNA expression levels of eight potential endogenous reference genes (Gapdh, Rn18s, Actb, Hprt, B2M, Rplp0, Gusb, Ctbp1). The expression stabilities of these reference genes were tested at different time points after MNU treatment using geNorm and NormFinder algorithms.
Results
A total of 65% of MNU-treated mice developed T-cell lymphomas, with the spleen and thymus as the major target organs. All candidate reference genes were amplified efficiently by quantitative reverse-transcription polymerase chain reaction (RT-qPCR). Gene stability evaluation after MNU treatment and during lymphomagenesis revealed that Ctbp1 and Rplp0 were the most stably expressed genes in the thymus and spleen, respectively. RT-PCR of thymus RNA using two additional sets of primer confirmed that Ctbp1 was the most stable of all the candidate reference genes.
Conclusions
We provided suitable endogenous controls for gene expression studies in the T-cell lymphoma model.
Similar content being viewed by others

Background
RT-qPCR is a powerful tool for quantifying gene expression and for validating results obtained by other techniques, such as microarray or RNA sequencing [1]. However, a suitable normalization method is necessary to detect variations in the expression levels of specific genes. Normalization usually involves selecting one or more so-called housekeeping genes as reference genes, such as Gapdh, Rn18s, or Actb [2,3,4]. However, some studies have reported extensive variations in the expression levels of putative reference genes among different tissues and stages of development, as well as in response to experimental treatments. For instance, Gapdh and Actb showed relatively unstable expression patterns in monosodium L-glutamate-induced obese mice [5], while Rn18s and Actb showed poor stability in colon cancer [6]. The precise evaluation of gene expression levels thus requires the selection of appropriate reference gene(s) for RT-qPCR analysis according to the particular experimental system.
T-cell lymphoma is an aggressive hematologic tumor resulting from the malignant transformation of T-cell progenitors [7]. Patients with T-cell lymphoma tend to present with very high circulating blast cell counts, mediastinal masses, and central nervous system involvement [8]. Despite a gradual increase in 5-year relapse-free survival rates following intensive chemotherapy, further advances in treatment outcomes require a better understanding of the mechanisms responsible for T-cell lymphoma [9]. T-cell lymphoma and B-cell precursor acute lymphoblastic leukemia have distinct clinical and laboratory features. Understanding the specific gene expression patterns may not only provide insights into the complex biological and pathological processes, but also help to predict disease and/or therapeutic treatment outcomes [10,11,12].
In the present study, we subjected a heterozygous p53-deficient mouse model (B6-Trp53tm1DAMR/NIFDC), established on a C57BL/6 background by embryonic stem (ES) cell targeting, to the intraperitoneal administration of N-methyl-N-nitrosourea (MNU). This model represents a valuable tool for the study of T-cell lymphoma. RT-PCR is a common method of monitoring changes in gene expression during tumor development, and a reference gene is needed to normalize the expression levels of other genes. To identify suitable reference genes during T-cell lymphoma development, we investigated the expression stabilities of eight commonly used candidate reference genes (Gapdh, Rn18s, Actb, Hprt, B2M, Rplp0, Gusb, and Ctbp1) by RT-qPCR at different time points following the administration of MNU.
Methods
Generation of p53 gene knockout mice and genotyping
A mouse p53 gene-targeting vector was constructed using a PGK promoter to drive the expression of a neomycin selection cassette (Neo). The targeting vector was introduced into C57BL/6 mouse ES cells by electroporation. After homologous recombination, the targeting vector replaced the p53 gene from exon 2 to 5. Neomycin resistant ES cell colonies were selected, screened by PCR, and injected into 151 wild-type BALB/c blastocysts. ES-cell-injected blastocysts were then transferred to 14 pseudo-pregnant mice and 8 chimeric mice were produced. Tail genomic DNA was isolated using a Tissue Genomic DNA Extraction Kit (Generay, Shanghai, China) and then subjected to PCR to verify deletion of the p53 gene. Genomic DNA of p53 deficient mice and wild-type mice were amplified with primer sets 1 (P53-WT-F, AGTTCTGCCACGTGGTTGGT; P53-WT-R, GTCTCCTGGCTCAGAGGGAG) or 2 (P53-WT-F, AGTTCTGCCACGTGGTTGGT; P53-Neo-R, CAGAGGCCACTTGTGTAGCG), with expected PCR products of 281 bp or 441 bp for wild-type and homozygous mutations, respectively. The male chimera mice were crossed with wild-type C57BL/6 female mice to generate heterozygous p53 gene knockout mice. For the heterozygous mutation, both bands were visible. C57BL/6 and BALB/c mice were produced in our breeding colony in Institute for Laboratory Animal Resources, National Institutes for Food and Drug Control (NIFDC). ES cell line used in this study was established from C57BL/6 mice in our lab. Blastocysts were obtained by standard protocol from BALB/c mice in our lab.
MNU-induced malignant lymphoma in p53 +/− mice
Fifty p53 +/−-deficient mice were divided into two groups and administered 37.5 mg/kg MNU or citrate buffer (control). MNU was dissolved in citrate-buffered saline and adjusted to pH 4.5 [13] before single intraperitoneal administration on day 1. Five mice from each group were sacrificed immediately and at 4, 8, and 12 weeks after the administration. Thymus and spleen, which were the main tumor target organs, were dissected for histopathological examination and RNA extraction.
Immunohistochemical analysis
Mouse tissues were fixed in 10% neutral buffered formalin, embedded in paraffin, and sectioned to about 5 μm and stained with hematoxylin and eosin (H&E) for histopathological examination.
Formalin-fixed, paraffin-embedded sections of thymus and spleen were processed for immunohistochemistry. Antibodies directed against CD3 (T-lymphocyte marker), CD20 (B-lymphocyte marker), and CD68 (macrophage marker) were used to classify the lineage of neoplastic cells in the thymus. Thymic malignant lymphoma or thymic sections for CD3, CD20, and CD68 staining were pretreated by incubation at 96 °C in Citra buffer (Zhongshan Golden Bridge Biocompany, Beijing, China) at pH 6 in a microwave for 10 min. Sections for CD3 staining were incubated with anti-CD3 antibody (clone LN10; Zhongshan Golden Bridge Biocompany), at 1:150 dilution, overnight at 4 °C after blocking with normal goat serum for 60 min at 37 °C. Sections for CD20 staining were incubated with anti-CD20 antibody (clone EP7; Zhongshan Golden Bridge Biocompany), at 1:200 dilution, overnight at 4 °C after blocking with normal goat serum for 60 min at 37 °C. Sections for CD68 staining were incubated with anti-CD68 antibody (clone PG-M1; Zhongshan Golden Bridge Biocompany), at 1:200 dilution, overnight at 4 °C after blocking with normal goat serum for 60 min at 37 °C. CD3, CD20, and CD68 immunoreactivities were all detected using a biotinylated rabbit anti-rat secondary antibody followed by an avidin-biotin-horseradish peroxidase complex, and visualized with diaminobenzidine. All immunohistochemical sections were counterstained with hematoxylin, dehydrated in graded concentrations of ethanol, and cover-slipped routinely using permanent mounting medium.
Tissue RNA extraction and cDNA synthesis
Spleen and thymus were dissected from the mice, immersed immediately in RNAlater stabilization reagent (Invitrogen, Carlsbad, CA, USA), and stored at −80 °C. Grinding of the tissues of three independent mice was performed in liquid nitrogen, followed by homogenization in TRIzol (Invitrogen). Total RNA was extracted in accordance with the manufacturer’s instructions. The amount of total RNA was determined by measuring the absorbances at 260 and 280 nm using a NanoDrop Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). All samples had an A260/A280 absorption ratio > 1.8. The RNA samples were reverse-transcribed to cDNA in a Reverse Transcription reaction mix using random hexamer primers, in accordance with the manufacturer’s instructions (Takara Bio Inc., Kusatsu, Japan).
Primer design and RT-qPCR
Primers for RT-qPCR assays of Gapdh, Rn18s, Actb, B2M, Hprt, Rplp0, Gusb, and Ctbp1 were designed using Primer Premier 5.0 (Table 1). Real-time PCR was performed using a Roche LightCycler 480 detection system (Roche Diagnostics, Germany). All standards and samples were run in triplicate in 96-well reaction plates. The cycle conditions were as follows: 15 s template denaturation at 95 °C and then 40 cycles of denaturation at 95 °C for 5 s and elongation at 60 °C for 30 s. This was followed by melting curve analysis, and baseline and cycle threshold values (Ct values) were determined automatically for all plates using Roche LightCycler 480 software.
Data analysis
The mRNA expression stability of each candidate gene was analyzed using the freely available Microsoft Excel-based software packages geNorm (https://genorm.cmgg.be/) and Norm-Finder (moma.dk/normfinder-software). Raw Ct values were transformed into relative quantities using the formula 2−∆ct. The obtained data were further analyzed using geNorm and NormFinder.
Results
Generation of T-cell lymphoma mouse model
MNU is a widely used genotoxic carcinogen [13, 14] used to induce T-cell lymphoma in various mouse models, including in the current study (Fig. 1). MNU-treated and control mice were observed twice a week and clinical signs were recorded until sacrifice. Moribund mice were necropsied at the earliest opportunity, and all surviving animals were sacrificed and necropsied at the end of 26 weeks. The thymus and spleen were dissected from the remaining mice for histopathological determination of lymphoma diagnosis and tumor frequency statistics. A total of 65% of MNU-treated mice developed lymphomas, compared with none of the control mice (Fig. 2a). No tumors other than malignant lymphoma were observed. The incidences of lymphomas in the two major target organs were 65% in the thymus and 50% in the spleen (Fig. 2b). Thymus and spleen sections from MNU-treated mice were subjected to hematoxylin and eosin staining (Fig. 2c, d), which showed effacement of thymic corticomedullary architecture by diffuse sheets of lymphoblasts with large euchromatic nuclei, as well as moderate to high numbers of and infiltration of lymphoblasts through the thymic capsule. Besides, immunostaining showed that all neoplastic cells in malignant lymphoma sections were positive for CD3 (Fig. 2e, f) and negative for CD20 (Fig. 2g, h) and CD68 (Fig. 2i, j), indicating that the malignant lymphomas were of T-lymphocyte origin.

Experimental design of analysis. Fifty p53 +/− mice were divided into two groups and administered 37.5 mg/kg MNU or citrate buffer. Five mice from each group were sacrificed immediately and at 4, 8, and 12 weeks after intraperitoneal injection. Thymus and spleen were dissected for RNA extraction. The most stable genes were determined in the MNU and control groups (Groups 1 and 2), and in mice grouped according to the time points after MNU administration (Groups A–D)

Features of lymphoma occurrence in p53-deficient heterozygous mice induced by 37.5 mg/kg MNU. a Tumor frequency in mice administered MNU or citrate buffer. b Tumor frequencies in thymus and spleen of mice administered MNU. c Hematoxylin and eosin (H&E) staining of lymphoma in thymus. d H&E staining of lymphoma in spleen. e, f. Thymus (e) and spleen (f) lymphoma stained positive for CD3 (T-lymphocyte marker). g, h Thymus (g) and spleen (h) lymphoma stained negative for CD20 (B-lymphocyte marker). i, j Thymus (i) and spleen (j) lymphoma stained negative for CD68 (macrophage marker). Magnification ×200, scale bar = 100 μm
Expression profiles of reference genes
We evaluated the expression stabilities of eight commonly used reference genes (Gapdh, Rn18s, Actb, B2M, Hprt1, Rplp0, Gusb, and Ctbp1) (Table 1) from different functional classes, to reduce the chance of coregulation of gene expression.
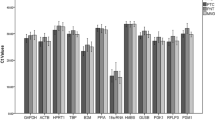
The primer sequences and sizes of the amplification fragments are shown in Table 2. Expression stability was assessed by RT-qPCR. The theoretical correlation coefficient (R2) for quality assays is 1, but is usually set as >0.980, representing the fit of tested samples to the regression line generated by the standard curve. All primer pairs showed an R2 of 0.992–0.999, indicating exponential template duplication. The amplification efficiencies for the eight reference genes ranged from 95 to 105% (Table 2), within the acceptable range of 90%–110% [1], indicating the suitability of the selected primers. Ct values are represented by box-and-whisker plots in Fig. 3. All reference genes displayed similar expression patterns in thymus and spleen, with wide variations in expression levels among different genes. The Rn 18S gene exhibited the lowest mean Ct values in thymus and spleen (10.7 and 11.7, respectively) and Gapdh exhibited the highest values (26.2 and 25.0, respectively).

Range of quantification cycle values of the candidate reference genes. Mean of Ct values for the eight reference genes in thymus (a) and spleen (b) with or without MNU treatment at each time point
Reference gene stability
We analyzed the data using geNorm and NormFinder to determine the stability of the genes and to identify the most suitable endogenous controls. geNorm and NormFinder are both examples of Microsoft Excel-based software [15]. geNorm analyzes each potential housekeeping gene by comparing its variation with that of all other evaluated reference genes. In contrast, NormFinder separately analyzes sample subgroups and takes into account intra- and intergroup variation for normalization factor calculations. Both algorithms calculate relative expression stability values for each reference gene, and the gene with the lowest stability value is considered the most stable gene. We collected tissues from MNU-treated and control mice at 0, 4, 8, and 12 weeks (Fig. 1) and analyzed expression levels by RT-qPCR at the various time points. Stability values were averaged among the four time points. NormFinder analysis revealed stability values of 0.104–0.918 in the thymus (Fig. 4a). The most stable reference gene in the thymus was Ctbp1, followed by Gusb and B2M, while Actb was determined as the least stable gene. geNorm analysis confirmed that Ctbp1 was the most stable reference gene and Actb was the least stable, consistent with the results of NormFinder (Fig. 4b). The stability values in the spleen analyzed by NormFinder were 0.287–1.181 (Fig. 4c). The most stable reference gene in the spleen was Rplp0, while Rn 18S was the least stable. The geNorm results were consistent with those of NormFinder (Fig. 4d). Owing to the different algorithms adopted by geNorm and NormFinder, the stability values produced by them could not be compared directly, so the candidate reference genes were ranked according to their stability values evaluated by geNorm and NormFinder (Table 3).

Expression stabilities of the eight candidate genes after administration of MNU. a, b Mean expression stability values in thymus from least to most stable are presented on the y- and x-axes using geNorm (a) and NormFinder (b). c, d Mean expression stability values in spleen from least to most stable expression are presented on the y- and x-axes using geNorm (c) and NormFinder (d)
The stabilities of genes during lymphomagenesis were also determined using NormFinder and geNorm. RT-qPCR data at each time point were grouped together and gene stability was analyzed across the time course. NormFinder analysis revealed stabilities of 0.169–0.873 in the thymus (Fig. 5a). The most stable reference gene was Ctbp1, followed by Gusb and Hprt, while Rn18s was the least stable. geNorm also identified Ctbp1 as the most stable reference gene and Actb as the least stable (Fig. 5b). Stability values in the spleen according to NormFinder ranged from 0.336 to 1.083 (Fig. 5c). The most stable reference gene in the spleen was Rplp0, while Rn18s was the least stable. The geNorm results were consistent with those of NormFinder (Fig. 5d).

Expression stabilities of the eight candidate genes during lymphoma development. a, b Mean expression stability values in thymus from least to most stable expression are presented on the y- and x-axes using geNorm (a) and NormFinder (b). c, d Mean expression stability values in spleen from least to most stable expression are presented on the y- and x-axes using geNorm (c) and NormFinder (d)
To rule out the possibility that the result was dependent on the specific primer used, two additional primers were designed for each reference gene. Sequence and amplification efficiencies are shown in Additional file 1: Table S1. RT-qPCR was then performed for thymus RNA using the additional primers, and mean Ct values of the primers at each time point are presented in Additional file 1: Figure S1. Raw Ct values were transformed into relative quantities using the formula 2−∆ct. The obtained data were further analyzed using geNorm and NormFinder. Consistent with our previous result, Ctbp1 was found to be the most stable gene both after MNU treatment (Additional file 1: Figure S2) and during lymphomagenesis (Additional file 1: Figure S3).
Discussion
The rapid increase in the incidence of T-cell lymphoma and its poor prognosis highlight the need for a reliable animal model to study the mechanisms by which this disease develops. We thus established a mouse T-cell lymphoma model by MNU induction. The current and previous reports indicate that the thymus and spleen are the target organs with the highest rates of lymphoma in such a model [13]. Monitoring changes in gene expression profiles during T-cell lymphoma development may provide clues to key genetic events, and may help to identify potential biomarkers for the diagnosis of T-cell lymphoma. RT-qPCR has been used widely to detect changes in gene expression because of its high accuracy and convenient methodology. However, it is essential to choose suitable reference genes for normalizing RT-qPCR data to ensure that the results reflect the true relative transcript abundances of genes within cells and tissues [16]. Although endogenous reference genes are widely used, few studies have examined the expression stability of such genes during tumorigeneses.
Previous studies evaluated the selection and effect of controls on normalized gene expression data; however, most of these involved human samples [17, 18]. We analyzed eight commonly used reference genes across T-cell lymphoma target tissues during different stages of tumorigenesis in an MNU-treatment animal model. The results of the current study indicated that the expression levels of so-called housekeeping genes were not stable, but were influenced by the stage of the lymphoma, tissue type, and MNU treatment. geNorm and NormFinder use different strategies to evaluate reference genes, and we therefore used both of these in the current study. Both analyses identified the same reference genes as the most stable after MNU induction and tumorigeneses. Ctbp1 and Rplp0 were selected as the best reference genes for the thymus and spleen, respectively, while Rn18s was considered to be the least stable gene. Other studies have also reported different stable genes in different tissues [19].
Ctbp plays central roles in both development and disease [20]. Ctbp1 and Ctbp2 are closely related genes that act as transcriptional corepressors. Ctbps primarily exert transcriptional repression through the recruitment of a corepressor complex to DNA. Ctbp overexpression has been observed in many human cancers, resulting in increased epithelial–mesenchymal transition, cancer cell survival, and stem cell-like features [21]. However, Ctbp1 exhibited a stable expression profile in the current T-cell lymphoma model. Further studies are needed to explore the precise functions of this gene.
Several programs are available for evaluating the stability of candidate genes, including GeNorm, NormFinder, and BestKeeper [15, 22, 23]. We increased the reliability of the results in the present study by using both GeNorm and NormFinder. The orders of stability of the less stable candidate reference genes were not completely consistent between NormFinder and geNorm, which could be explained by the different principles that they use. The model-based approach used by NormFinder has the advantage of being able to differentiate between intragroup and intergroup variation, making it a suitable tool for identifying candidate genes when different sample groups are assessed. However, it has the disadvantage of requiring larger sample sizes than geNorm (>8). In contrast, the pairwise correlation used by the geNorm algorithm is known to be a strong algorithm for small sample sizes.
Conclusions
In conclusion, we identified Ctbp1 and Rplp0 as the best reference genes for thymus and spleen, respectively, in an MNU-induced T-cell lymphoma mouse model. To the best of our knowledge, this study provides the first systemic evaluation of reference genes in a mouse model of lymphoma.
Abbreviations
- Ct values:
-
Cycle threshold values
- ES:
-
Embryonic stem
- H&E:
-
Hematoxylin and eosin
- MNU:
-
N-methyl-N-nitrosourea
- NIFDC:
-
National Institutes for Food and Drug Control
- PCR:
-
Real-time polymerase chain reaction
- RT-qPCR:
-
Quantitative reverse-transcription polymerase chain reaction
References
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–22.
de Jonge HJ, Fehrmann RS, de Bont ES, Hofstra RM, Gerbens F, Kamps WA, de Vries EG, van der Zee AG, te Meerman GJ, ter Elst A. Evidence based selection of housekeeping genes. PLoS One. 2007;2(9):e898.
Huggett J, Dheda K, Bustin S, Zumla A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005;6(4):279–84.
Radonic A, Thulke S, Mackay IM, Landt O, Siegert W, Nitsche A. Guideline to reference gene selection for quantitative real-time PCR. Biochem Biophys Res Commun. 2004;313(4):856–62.
Matouskova P, Bartikova H, Bousova I, Hanusova V, Szotakova B, Skalova L. Reference genes for real-time PCR quantification of messenger RNAs and microRNAs in mouse model of obesity. PLoS One. 2014;9(1):e86033.
Sorby LA, Andersen SN, Bukholm IR, Jacobsen MB. Evaluation of suitable reference genes for normalization of real-time reverse transcription PCR analysis in colon cancer. J Exp Clin Cancer Res. 2010;29:144.
Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350(15):1535–48.
Graux C, Cools J, Michaux L, Vandenberghe P, Hagemeijer A. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from thymocyte to lymphoblast. Leukemia. 2006;20(9):1496–510.
Meijerink JP. Genetic rearrangements in relation to immunophenotype and outcome in T-cell acute lymphoblastic leukaemia. Best Pract Res Clin Haematol. 2010;23(3):307–18.
Ge X, Yamamoto S, Tsutsumi S, Midorikawa Y, Ihara S, Wang SM, Aburatani H. Interpreting expression profiles of cancers by genome-wide survey of breadth of expression in normal tissues. Genomics. 2005;86(2):127–41.
Patsialou A, Wang Y, Lin J, Whitney K, Goswami S, Kenny PA, Condeelis JS. Selective gene-expression profiling of migratory tumor cells in vivo predicts clinical outcome in breast cancer patients. Breast Cancer Res. 2012;14(5):R139.
van’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–6.
Morton D, Bailey KL, Stout CL, Weaver RJ, White KA, Lorenzen MJ, Ball DJ. N-Methyl-N-Nitrosourea (MNU): a positive control chemical for p53+/− mouse carcinogenicity studies. Toxicol Pathol. 2008;36(7):926–31.
Takaoka M, Sehata S, Maejima T, Imai T, Torii M, Satoh H, Toyosawa K, Tanakamaru ZY, Adachi T, Hisada S, Ueda M, Ogasawara H, Matsumoto M, Kobayashi K, Mutai M, Usui T. Interlaboratory comparison of short-term carcinogenicity studies using CB6F1-rasH2 transgenic mice. Toxicol Pathol. 2003;31(2):191–9.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034.
Kozera B, Rapacz M. Reference genes in real-time PCR. J Appl Genet. 2013;54(4):391–406.
de Kok JB, Roelofs RW, Giesendorf BA, Pennings JL, Waas ET, Feuth T, Swinkels DW, Span PN. Normalization of gene expression measurements in tumor tissues: comparison of 13 endogenous control genes. Lab Investig. 2005;85(1):154–9.
Janssens N, Janicot M, Perera T, Bakker A. Housekeeping genes as internal standards in cancer research. Mol Diagn. 2004;8(2):107–13.
Svingen T, Letting H, Hadrup N, Hass U, Vinggaard AM. Selection of reference genes for quantitative RT-PCR (RT-qPCR) analysis of rat tissues under physiological and toxicological conditions. Peer J. 2015;3:e855.
Stankiewicz TR, Gray JJ, Winter AN, Linseman DA. C-terminal binding proteins: central players in development and disease. Biomol Concepts. 2014;5(6):489–511.
Blevins MA, Kouznetsova J, Krueger AB, King R, Griner LM, Hu X, Southall N, Marugan JJ, Zhang Q, Ferrer M, Zhao R. Small molecule, NSC95397, inhibits the CtBP1-protein partner interaction and CtBP1-mediated transcriptional repression. J Biomol Screen. 2015;20(5):663–72.
Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64(15):5245–50.
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26(6):509–15.
Acknowledgments
We thank Tom Buckle, MSc, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Funding
This work is supported by National Natural Science Foundation of China (Grant number: 81,502,396 to Xi Wu).
Availability of data and materials
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
CF designed the research. XW, SL, JL, SZ, YY, CW, WG and QZ performed the research and wrote part of the results. XW, SL, BL and CF analyzed the data. XW and CF wrote the main paper. All authors discussed the results and implications and commented on the manuscript at all stages. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Mice used in this study were housed and handled strictly in accordance with the institutional (National Institutes for Food and Drug Control) guidelines for animal care and use. The study protocol was approved by the NIFDC Institutional Animal Care and Use Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Stability analysis of reference genes in thymus by two additional primers. To rule out the possibility that the result was dependent on the specific primer used, two additional primers were designed for each reference gene. Sequence and amplification efficiencies, mean Ct values of the primers at each time point and result analyzed using geNorm and NormFinder were presented in the file. Figure S1. Range of quantification cycle values of the candidate reference genes. Mean of Ct values of primer 2 (A) and primer 3 (B) for the eight reference genes in thymus with or without MNU treatment at each time point. Figure S2. Expression stabilities of the eight candidate genes after MNU treatment in thymus. A, B. Mean expression stability values in thymus from least to most stable are presented on the y- and x-axes using geNorm (A) and NormFinder (B). Figure S3. Expression stabilities of the eight candidate genes during lymphoma development in thymus. A, B. Mean expression stability values in thymus from least to most stable expression are presented on the y- and x-axes using geNorm (A) and NormFinder (B). (DOCX 413 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wu, X., Liu, S., Lyu, J. et al. Endogenous controls of gene expression in N-methyl-N-nitrosourea-induced T-cell lymphoma in p53-deficient mice. BMC Cancer 17, 545 (2017). https://doi.org/10.1186/s12885-017-3536-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-017-3536-6