
Abstract
Alzheimer’s disease (AD) is a primary cause of dementia. The complement system is closely related to AD pathology and may be a potential target for the prevention and treatment of AD. In our study, we conducted a bioinformatics analysis to analyze the role of the complement system and its related factors in AD using Gene Expression Omnibus (GEO) data. We also conducted a functional analysis. Our study verified that 23 genes were closely related to differentially expressed complement system genes in diseases after intersecting the disease-related complement system module genes and differentially expressed genes. The STRING database was used to predict the interactions between the modular gene proteins of the differential complement system. A total of 21 gene proteins and 44 interaction pairs showed close interactions. We screened key genes and created a diagnostic model. The predictive effect of the model was constructed using GSE5281 and our study indicated that the predictive effect of the model was good. Our study also showed enriched negative regulation of Notch signaling, cytokine secretion involved in the immune response pathway, and cytokine secretion involved in immune response hormone-mediated apoptotic signaling pathway. We hope that our study provides a promising target to prevent and delay the onset, diagnosis, and treatment of AD.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is a primary cause of dementia. The number of patients with AD is expected to reach 100 million by 2050. AD has become one of the most expensive, deadliest, and burdensome diseases of this century [1]. Therefore, it is important to understand the pathogenesis of AD and identify effective treatment methods.
At present, there are many hypotheses about the pathogenesis of AD such as: β-amyloid protein (Aβ) hypothesis, tau protein hypothesis, central cholinergic injury hypothesis, excitatory amino acid toxicity hypothesis, and neuroimmunity hypothesis. Neural immunity has been proven to be closely related to the pathology of AD, and its important components: immune cells (such as T cells [2] and B cells [3]), and various immune active substances (such as complement system-related molecules [4] and neuroinflammatory factors [5]), have been reported to be involved in the pathogenesis of AD. The complement system is an important part of the neuroimmune system and participates in multiple pathological processes in AD. C3 [6], complement receptor 1 (CR1) [7], C1q [8], and C3aR [6] are involved in Aβ metabolism. C4 [9], C3 [10, 11], and CR1 [12] were involved in tau pathology. C1q [13] and CR1 [12] are involved in neuroinflammation. C1q [8], C3 [8], and CR1 [12] are involved in synaptic formation. In conclusion, the complement system is closely related to AD pathology and may be a potential target for the prevention and treatment of AD.
As we all know, many factors are also involved in the complement system to regulate the pathological process of AD. Examining literatures from these years, we have found that the complement system can modulate nucleotide binding oligomerization domain leucine rich repeat and pyrin domain containing 3 inflammasome [14], triggering receptor expressed on myeloid cells 2 [15], and several inflammation-related factors such as tumor necrosis factor [16, 17], interleukin-1 [16, 17], interleukin-6 [16, 17], C-reactive protein [17], and interleukin-10 [16]. The above research suggests that the complement system may be involved in the pathology of AD through its associated genes, proteins, and related signaling pathway. However, there is currently no literature reporting on this aspect. Hence, in the present study, we focused on identifying the role of the complement system and its key related genes, proteins and signaling pathways in AD. We conducted a bioinformatics analysis to analyze the role of the complement system and its related factors in AD. Moreover, functional enrichment analyses and a diagnostic prediction model were also carried out in our study. We hope that our study provides a promising target to prevent and delay the onset, diagnosis, and treatment of AD.
Methods
Data acquisition
The datasets generated and analyzed in the current study are available in the NCBI GEO repository (http://www.ncbi.nlm.nih.gov/geo/) under the accession numbers (GSE109887 and GSE5281). The dataset originated from the middle temporal gyrus tissue. Our primary criterion for sample selection was the dataset sample size, followed by gene quantity in the gene expression matrix of the dataset. Through GEO database retrieval, we identified that GSE109887 had the largest sample size from middle temporal gyrus tissue and a more abundant gene quantity in its expression matrix. Consequently, we designated it as the primary dataset for analysis (training set); with GSE5281, featuring fewer samples and genes, serving as the validation set. Both GSE109887 and GSE5281 are datasets used for Expression Profiling by Array [18]. Data on AD patients data are available from the NCBI GEO database [19] (http://www.ncbi.nlm.nih.gov/geo/), and the serial number of the AD patients’ dataset is GSE109887. The data included samples of middle temporal gyrus tissue from 46 patients with AD and 32 healthy controls without AD. The gene microarray was determined using the GPL10904 Illumina HumanHT-12 V4.0 expression bead-chip (gene symbol) platform. This dataset was primarily treated as an analytical dataset.
In addition, we downloaded the AD patients’ validation dataset from the NCBI GEO database. The serial number of the AD patient data was GSE5281. The data included samples of middle temporal gyrus tissue from 16 patients with AD and 12 healthy controls without AD. Gene microarray analysis was performed using the GPL570 (HG-U133_Plus_2) Affymetrix Human Genome U133 Plus 2.0 Array. This dataset was primarily treated as an analytical dataset to validate the diagnostic models.
When conducting data analysis, we performed an analysis of abnormal/outlier samples on the training set data. As shown in the figure below, all samples, as represented by box plots and PCA analysis charts, indicate that the analyzed data set does not contain any abnormal samples (Supplementary Fig. 1).
Data preprocessing
For the above two datasets, firstly, we downloaded preprocessed, standardized, and log2-transformed probe expression matrices from the GEO database. Secondly, we obtained annotation files for the platforms and conducted a one-to-one matching between probe numbers and Gene symbols. Probes that did not have a matching Gene symbol were excluded. In cases where different probes mapped to the same gene, we calculated the mean expression value of these different probes to represent the final expression value for that gene, which was utilized in subsequent analyses. After preprocessing, GSE109887 yielded an expression matrix of 31,700 genes across 78 samples; whereas GSE5281 produced an expression matrix of 23,520 genes across 28 samples.
Differential gene analysis
Based on the analysis dataset, we employed the limma package [20] (Version 3.10.3, http://www.bioconductor.org/packages/2.9/bioc/html/limma.html) and its classical Bayesian methods to analyze differential gene expression between AD and Control groups. All genes underwent analysis, resulting in corresponding P values and logFC values. Additionally, the BH correction was applied to obtain adjusted P-values (adj.p.val). We evaluated at two levels: differential fold change and significance. The threshold for differential expression was set as adj.p.val < 0.05 and |logFC|> 0.585. After identifying differentially expressed genes, a volcano plot was generated, and expression distribution heatmaps between normal and disease groups were separately illustrated [21,22,23]. After identifying differentially expressed genes, a volcano plot was generated, and expression distribution heatmaps between normal and disease groups were separately illustrated.
WGCNA (Weighted Gene Co-expression Network analysis) was used to analyze disease-associated complement system related gene modules
We conducted a search in the CTD database [24] (https://ctdbase.org/) using the keyword “complement system” to retrieve genes related to the complement system. In addition, Additionally, a similar search was performed in GeneCards [25] (https://www.genecards.org/) using the keyword “complement system”, with a threshold set at Relevance score >= 10 [26, 27], to obtain complement system-related genes. The genes obtained from both databases were merged, removing duplicates, and then matched with the genes in the analysis dataset. The resulting expression matrix of complement system genes was utilized for WGCNA.
We utilized the R package WGCNA [28] (Version 1.61, https://cran.r-project.org/web/packages/WGCNA/) to analyze the complement system genes, aiming to identify modules of genes that exhibit high levels of co-expression variation.
In the WGCNA algorithm, the elements of the gene co-expression matrix are weighted values representing the correlation coefficients between genes. The criterion for selecting these weights is to ensure that the connections between genes within each gene network follow a scale-free network distribution. The specific weighted values are referred to as the soft power. Initially, we set a series of powers and then calculated the square value of the correlation coefficient between connectivity k, p (k), and the average connectivity under each power value. An appropriate power value is then selected to achieve a scale-free network distribution among genes in the network. Subsequently, parameters were set based on clustering and dynamic pruning (minModuleSize = 30: each module contained at least 30 genes; MEDissThres = 0.25: combining modules whose similarity degree was greater than 0.75). Genes with a high correlation were clustered into modules, and the correlation between modules and phenotypes was calculated. Here, the phenotype refers to whether the samples were diseased or normal. Modules with absolute correlation coefficients greater than 0.3 and significance values less than 0.05 were selected as modules closely related to diseases.
Acquisition of disease-related differential complement system genes
Taking the intersection of the disease-related complement system module genes (method 2.4) and differential genes (method 2.3) obtained above, further identify differential complement system genes closely associated with the disease.
Interaction network analysis and co-expression relationship analysis of corresponding proteins of genes in the differential complement system
In order to understand the protein-protein interaction (PPI) relationships among the differentially expressed complement system genes obtained above, we utilized the online STRING database [29] (Version: 11.0, http://www.string-db.org/) for predicting and analyzing potential interactions between the proteins encoded by these genes. The species was set to Homo sapiens (homo), and the PPI score threshold was set to 0.4 (medium confidence).
In addition, Pearson’s correlation coefficients were calculated between gene pairs in the differential complement system to observe the relationships between gene co-expression trends.
Key gene screening and diagnostic model construction
Based on the differential complement system genes obtained above, we first calculated the diagnostic AUC value of each gene using the expression value of each gene in each sample, combined with the grouping information of the sample, and selected the gene with an AUC > 0.8 as the candidate gene related to the disease. Subsequently, the LASSO (least absolute shrinkage and selection operator) algorithm was used to screen the feature genes. Glmnet package Version 4.0-2 using R language 3.6.1 [20] (https://cran.r-project.org/web/packages/glmnet/index.html) for a preliminary screening of candidate genes associated with disease regression analysis The parameter was set to nfold = 20, that is, 20-fold cross-validation was carried out to screen characteristic genes, and the following formula was used to construct the model:
Here, βgene represents the LASSO regression coefficient of each gene, and Expgene represents gene expression level in each sample.
After the risk score was obtained, we considered the median value as the critical value and divided the samples into HighRisk and LowRisk groups for subsequent analysis.
Furthermore, to validate whether the model has diagnostic value, GSE5281 was utilized. Firstly, the expression values of the model genes were extracted from each sample. Subsequently, in conjunction with the LASSO regression coefficients obtained earlier, the same formula was employed for model construction. ROC curves were then plotted by combining the sample grouping information.
Comparison of immune microenvironment between HighRisk group and LowRisk group
We downloaded 17 immune gene sets from the Import database (https://www.immport.org/home) [30] using the R package GSVA [31] (version: 1.36.2, http://bioconductor.org/packages/release/bioc/html/GSVA.html), which is based on single-sample enrichment analysis (ssGSEA). We obtained significant differential P values between the high- and low-risk groups using the Wilcoxon test. The Spearman correlation coefficients of each gene and the differential immune gene set in the model were calculated, and a correlation heat map was drawn.
In order to understand AD-like group and normal-like immune cells infiltrating differences between groups, here we use the R package MCPcounter (https://github.com/ebecht/MCPcounter) [32] algorithm based on the analysis of the expression level in the data set. The infiltration levels of eight types of immune cells (T cells, CD8 + T cells, cytotoxic lymphocytes, B lineage, NK cells, monocyte lineage, myeloid dendritic cells, and neutrophils) and two types of stromal cells (endothelial cells and fibroblasts) were calculated. Wilcoxon tests were used to compute the significance of differences in the infiltration levels of various immune or stromal cells between the HighRisk and LowRisk groups. Furthermore, we calculated Spearman correlation coefficients between individual genes in the model and differential immune cell infiltrations, and a correlation heatmap was generated to visualize these relationships.
In addition, the Wilcoxon test was used to compare differences in HLA family genes between the AD-like group and normal-like groups. The Pearson correlation coefficient between each gene and the HLA family gene in the model was calculated to construct a correlation heat map.
Pathway enrichment analysis of the HighRisk and LowRisk groups
A total of 51 HALLMARK gene sets were downloaded from MSigDB v7.1 [33] (http://software.broadinstitute.org/gsea/msigdb/index.jsp), and these genes were used as the background for enrichment analysis. The enrichment scores of each HALLMARK pathway in each sample were calculated based on the expression matrix of the analysis data set by the GSVA (gene set variation analysis) algorithm. A scoring matrix was obtained using the GSVA R-packet. Then, a difference analysis of the HighRisk group vs. the LowRisk group was carried out for each pathway using the R packet limma. The results were considered as significant pathways after screening “BH” adjusted (adj. P value < 0.05 and |t value|> 2).
GO BP and KEGG enrichment analysis of key model genes
The key model (in process) and KEGG pathway enrichment analyses were performed using the Cytoscape Software (version 3.4.0, http://chianti.ucsd.edu/cytoscape-3.4.0/) [34] with the Cluego plugin [35] (version 2.5.9, https://apps.cytoscape.org/apps/cluego). The significance threshold was set at a P value < 0.05. The Cytoscape software was used to visualize the enrichment results.
Results
Data preprocessing and difference analysis
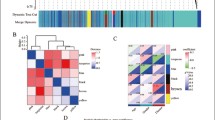
Using the above method, two sets of data were downloaded, preprocessed, and annotated. According to the threshold value, 187 upregulated and 280 downregulated genes were identified. For more details, see Supplementary Table 1. Differential volcanic and thermal maps are shown in Fig. 1A and B.

Data preprocessing and Difference analysis. A The differential volcanic map; B the thermal map
WGCNA (Weighted Gene Co-expression Network analysis) was used to analyze disease-associated complement system related gene modules
Based on a previously described method, first a total of 760 gene expression matrices related to the complementation system were matched. We then chose six as the soft threshold in the WGCNA analysis according to Fig. 2A and B. Second, based on clustering and dynamic pruning methods- high-correlation genes were aggregated into modules; which were then clustered. The modules with correlation coefficients greater than 0.75, that is, the modules with dissimilarity coefficients less than 0.25; were merged and finally integrated into four modules (gray indicates genes with less than any enriched module) (Fig. 2C and D).

WGCNA of disease-associated complement system related gene. A The relationship between scale-free topology model fit and soft-threshold power; B The relationship between mean connectivity and soft-threshold power; C Clustering of module eigengences; D Cluster of dendrogram
Furthermore, by calculating the correlation between the feature vector gene of each module (the feature vector gene is the first principal component gene E of a specific module, representing the overall level of gene expression in the module) and the phenotype (whether the sample is OA or Normal), as shown in Fig. 3, it can be seen that the turquoise module (162 genes; correlation coefficient r = 0.51 and P Value < 0.01) had the most significant positive correlation with AD; and the blue module (102 genes; correlation coefficient r = 0.33, P Value = 0.003) also showed a significant positive correlation with AD. The yellow module (31 genes; correlation coefficient r = 0.43 and P < 0.01) was significantly negatively correlated with AD.

The correlation between the feature vector gene of each module
Acquisition of disease-related differential complement system genes
A total of 23 genes were confirmed to be closely related to the differential complement system genes of diseases after intersecting the disease-related complement system module genes and differential genes. As shown in Fig. 4, 18 genes (GFAP, PDGFRB, NFKBIA, TNFRSF1B, NOTCH1, BCL6, CSF1R, LEP, DCLRE1C, KCNJ10, MP2K1, VIP, SNCA, ENO2, SST, UCHL1, HPRT1, and STAT4) belonged to the turquoise module; three genes (CD14, ITGB2, and SPP1) belonged to the blue module; and two genes (STX1A and SYP) belonged to the yellow module. These three modules were deemed as pivotal module genes associated with AD.

A total of 23 genes were confirmed closely related differential complement system genes of diseases after intersected of disease-related complement system module genes and differential genes
Protein interaction network and correlation analysis
The STRING database was used to predict the interactions between the modular gene proteins of the differential complement system. As shown in Fig. 5A, 21 genes and 44 interaction pairs showed close interactions. We further calculated the Pearson correlation coefficient between the genes and significant P-values for the 23 differences in the complement system. Except for SPP1, the other genes had close and significant positive or negative relationships (Fig. 5B). The results of PPI analysis suggest potential interactions between the complement system and various related proteins, providing new directions for our future exploration of the role of the complement system in AD.

Protein interaction network and correlation analysis. A PPI network. There were 21 genes; B the Pearson correlation coefficient between the gene and significant P value values in the 23 differences of the complement system
Key gene screening and diagnostic model construction
Using the method described above, 23 genes related to the complement system were identified. The diagnostic AUC value was calculated by combining it with the sample grouping. Eleven genes with an AUC > 0.8 were obtained (which were initially used as disease-related candidate genes) and were identified.
Further, LASSO regression was performed for these 11 genes- and the results are shown in Fig. 6A-B. Six key characteristic genes, namely NFKBIA, TNFRSF1B, BCL6, KCNJ10, VIP, and SST, were identified.

Key gene screening and diagnostic model construction. A-B Six key characteristic genes NFKBIA, TNFRSF1B, BCL6, KCNJ10, VIP and SST were obtained. C-D RiskScore model was constructed by combining the above 6 genes and corresponding regression coefficients. E-F The prediction effect of the model
According to the method described above, the RiskScore model was constructed by combining the above six genes and their corresponding regression coefficients. The results are shown in Fig. 6C-D. According to the ROC curve, the area under the curve exceeded 0.85: indicating that this model had a good disease prediction effect.
To verify the prediction effect of the model, it was reconstructed in GSE5281 according to this same method- and the results are shown in Fig. 6E-F. The area under the curve (AUC) is above 0.9, which is consistent with previous conclusions, indicating that the prediction effect of the model is good.
Comparison of immune microenvironment between HighRisk group and LowRisk group
According to the method described above, 17 types of immune gene sets were analyzed in accordance with the method described for the HighRisk and LowRisk group comparisons. The results showed that a total of 14 immune gene sets showed significant differences between high and low-risk groups, as shown in Fig. 7A. For this purpose, a correlation heat map was drawn between the model genes and the 14 different immune gene sets as shown in Fig. 7B. The relationships with the largest positive and negative correlation coefficients were selected for display, as shown in Fig. 7C-D.

Comparison of immune microenvironment between HighRisk group and LowRisk group. A The results showed that a total of 14 immune gene sets showed significant differences between high and low risk groups. B The correlation heat map between model genes and 14 different immune gene sets was drawn. C-D The relationship with the largest positive and negative correlation coefficients was selected for display; E 7 kinds of immune cells showed significant differences; F The heat map of the correlation between model genes and differential immune cells; G-H The relationship with the largest positive and negative correlation coefficients was selected for display; I The differences of HLA family genes between the HighRisk group and the LowRisk group; J 9 genes showed significant differences. J The correlation heat map of model genes and different HLA family genes; K-L The relationship with the largest positive correlation coefficient and negative correlation coefficient was selected to display
Furthermore, the immune and stromal cells in the high- and low-risk groups were compared. As shown in Fig. 7E, seven types of immune cells showed significant differences; except for NK cells in the high-risk group, which were lower than those in the low-risk group. The other 6 kinds of cells were significantly higher in the high-risk group than in the low-risk group. A heat map of the correlation between the model genes and differentially expressed immune cells is shown in Fig. 7F. The relationships with the largest positive and negative correlation coefficients were selected for display, as shown in Fig. 7G-H.
According to the method described above, we also compared the differences in HLA family genes between the high- and low-risk groups, as shown in Fig. 7I. Fifteen and nine genes showed significant differences, respectively. A correlation heat map of the model genes and different HLA family genes was drawn, as shown in Fig. 7J. The relationship with the largest positive and negative correlation coefficients was selected for display, as shown in Fig. 7K-L. The results of this section suggest that the complement system may be involved in the pathological mechanisms of AD through interactions with immune-related molecules. This points towards future research directions of AD.
Pathway enrichment analysis in high-risk and low-risk groups
The enrichment scores of each HALLMARK path in each sample were calculated using the GSVA algorithm. According to the method threshold, 15 HALLMARK pathways in HighRisk and 19 low-risk HALLMARK pathways were significantly enriched; as shown in Fig. 8.

Pathway enrichment analysis in high-risk and low-risk groups
GO BP and KEGG enrichment analysis of key model genes
A total of 67 GO BP were enriched in Fig. 9A); however, no KEGG pathways were enriched. Mainly enriched negative regulation of Notch signaling, cytokine secretion involved in the immune response, hormone-mediated apoptotic signaling pathway, and other key functions are shown in Fig. 9B. These studies suggest that the complement system may be involved in the pathological processes of AD through the above signaling pathways. However, the specific mechanisms require further validation.

GO BP and KEGG enrichment analysis of key model genes. A A total of 67 GO BP; B Other key functions
Discussion
In this study, the gene information related to AD was analyzed using biogenic analysis. We also identified differences in the regulation of complement-system-related proteins associated with them. A model was constructed to predict the molecular characteristics of AD. Moreover, the relationship between the complement system and AD affecting immune-related genes and inflammatory factors is discussed. This is the first biogenic analysis to comprehensively explore the roles and possible mechanisms of the complement system in AD.
Late-onset AD (LOAD), which accounts for approximately 95% of AD cases, is a multifactorial disease with a heritability of over 58% [36]. Since 2009, large genome-wide association studies (GWAS) have identified more than 75 independent genetic risk factors for LOAD [37,38,39,40]. Several studies have been conducted to explore the link between complement system genes and AD. Among the GWAS statistically significant (GWS) hits are two genes encoding proteins of the complement pathway: CR1 encoding the membrane protein complement receptor 1 (CR1) and CLU encoding the plasma regulator clusterin [38]. CR1 and CLU are among the most significant GWAS hits, ranking high in the top 10. These strong associations provide the impetus for this review of complement genetics in LOAD. Our study identified several complement system-related genes in AD. Among the 23 genes previous studies have shown their correlation with AD, we have: GFAP [41, 42], PDGFRβ [43,44,45], NFKBIA [46], TNFRSF1B [47], NOTCH1 [48], BCL6 [49], CSF1R [50], LEP [51], DCLRE1C [52], KCNJ10 [53], MAP2K1 [54], VIP [55], SNCA [56], ENO2 [57], SST [58], UCHL1 [59], HPRT1 [60], STAT4 [61], CD14 [62], ITGB2 [63], SPP1 [64], STX1A [65], and SYP [66]. And these genes are involved in the regulation of molecules associated with the complement system, for instance, GFAP [67], PDGFRβ [68], NFKBIA [69], TNFRSF1B [70], NOTCH1 [71], BCL6 [72], CSF1R [73], LEP [74], DCLRE1C [75], KCNJ10 [76], MAP2K1 [77], VIP [78], SNCA [79], ENO2 [80], SST [81], UCHL1 [82], HPRT1 [83], STAT4 [84], CD14 [85], ITGB2 [86], SPP1 [87], STX1A [88], and SYP [89]. However, few studies have discussed their role in AD pathology by modulating the complement system, our study may be a new perspective for our future exploration of the relationship between AD and the complement system. A systematic discussion of the relationship among these genes, the complement system, and AD is likely to become one of the directions for future AD research. The above results show that these proteins are interrelated, and further research is needed to understand their mechanisms. Our research revealed enrichment in the negative regulation of the Notch signaling pathway, cytokine secretion associated with the immune response pathway, and the hormone-mediated apoptotic signaling pathway. These signaling pathway may play a vital role in AD pathology in conjunction with the complement system. However, further experiments are required to investigate this hypothesis.
It is well known that AD lacks effective diagnostic measures. Hence, it may be worthwhile looking into potential brain complement specific biomarkers which can indicate the susceptibility of AD. The classic AD biomarkers are core CSF biomarkers: Aß, t-tau, and phosphorylated tau protein (p-tau), and PET imaging of glucose metabolism and amyloid deposition. However, some patients with early-stage AD cannot be diagnosed using these means [90]. Our findings propose that the diagnostic potential for AD could be enhanced through the consideration of a set of six key genes (NFKBIA, TNFRSF1B, BCL6, KCNJ10, VIP, and SST). When we queried recent literature, we found that NFKBIA was associated with AD in some articles [91,92,93,94,95]. The relationship between the TNFRSF1B variant rs976881 and the levels of soluble TNFR2 in the cerebrospinal fluid influences various markers of AD severity and cognitive domains [47]. It is reportedly achieved by targeting BCL6, which presents a promising avenue to explore for the prevention and treatment of Aβ-induced neuronal damage in AD [96]. Moreover, genetic screening revealed that mutations in KCNJ10 play a significant role in neurodegenerative disorders such as AD [53]. VIP may significantly boost microglial uptake of fibrillar Aβ42 and this heightened phagocytic function relied on the activation of the Protein kinase C signaling pathway in AD pathology [97]; VIP was also reported to be involved in Aβ accumulation in AD [98]. A study pointed out an Interaction between Aβ and SST [58]. The validation experiments further affirmed the robust prediction accuracy of our model. It suggests that the model might serve as a diagnostic tool for AD by incorporating classic AD biomarkers and screening for these identified key genes.
Conclusions
A total of 21 gene proteins and 44 interaction pairs showed close interactions. We screened key genes and created a diagnostic model. The predictive effect of the model was constructed using GSE5281 and our study indicated that the predictive effect of the model was good. Our study also showed enriched negative regulation of Notch signaling, cytokine secretion involved in the immune response pathway and hormone-mediated apoptotic signaling pathway. In this study, key complement system-related genes with good diagnostic value were screened through bioinformatics analysis, which provided some clues for a better understanding of the potential molecular mechanisms of AD. The main limitation of our study is that it primarily involves bioinformatics analysis, lacking relevant clinical or further experimental validation. Hence, these findings of our study must be verified in future studies.
Availability of data and materials
All data during the current study are available from the corresponding author on reasonable request.
References
Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE, et al. Alzheimer’s disease. Lancet. 2021;397(10284):1577–90. https://doi.org/10.1016/S0140-6736(20)32205-4.
Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature. 2020;577(7790):399–404. https://doi.org/10.1038/s41586-019-1895-7.
Ahn JJ, Abu-Rub M, Miller RH. B cells in neuroinflammation: new perspectives and mechanistic insights. Cells. 2021;10(7):1605. https://doi.org/10.3390/cells10071605.
Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell. 2020;182(4):976-91.e19. https://doi.org/10.1016/j.cell.2020.06.038.
Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, et al. LC3-associated endocytosis facilitates beta-amyloid clearance and mitigates neurodegeneration in murine Alzheimer’s disease. Cell. 2019;178(3):536-51.e14. https://doi.org/10.1016/j.cell.2019.05.056.
Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J Neurosci. 2016;36(2):577–89. https://doi.org/10.1523/JNEUROSCI.2117-15.2016.
Zhu XC, Dai WZ, Ma T. Impacts of CR1 genetic variants on cerebrospinal fluid and neuroimaging biomarkers in Alzheimer’s disease. BMC Med Genet. 2020;21(1):181. https://doi.org/10.1186/s12881-020-01114-x.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–6. https://doi.org/10.1126/science.aad8373.
Panitch R, Hu J, Chung J, Zhu C, Meng G, Xia W, et al. Integrative brain transcriptome analysis links complement component 4 and HSPA2 to the APOE epsilon2 protective effect in Alzheimer disease. Mol Psychiatry. 2021;26(10):6054–64. https://doi.org/10.1038/s41380-021-01266-z.
Hu J, Yang Y, Wang M, Yao Y, Chang Y, He Q, et al. Complement C3a receptor antagonist attenuates tau hyperphosphorylation via glycogen synthase kinase 3beta signaling pathways. Eur J Pharmacol. 2019;850:135–40. https://doi.org/10.1016/j.ejphar.2019.02.020.
Litvinchuk A, Wan YW, Swartzlander DB, Chen F, Cole A, Propson NE, et al. Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer’s disease. Neuron. 2018;100(6):1337-53.e5. https://doi.org/10.1016/j.neuron.2018.10.031.
Zhu XC, Liu L, Dai WZ, Ma T. Crry silencing alleviates Alzheimer’s disease injury by regulating neuroinflammatory cytokines and the complement system. Neural Regen Res. 2022;17(8):1841–9. https://doi.org/10.4103/1673-5374.332160.
Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019;25(3):496–506. https://doi.org/10.1038/s41591-018-0336-8.
Yang J, Wise L, Fukuchi KI. TLR4 cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer’s disease. Front Immunol. 2020;11:724. https://doi.org/10.3389/fimmu.2020.00724.
Zhong L, Sheng X, Wang W, Li Y, Zhuo R, Wang K, et al. TREM2 receptor protects against complement-mediated synaptic loss by binding to complement C1q during neurodegeneration. Immunity. 2023;56(8):1794-808.e8. https://doi.org/10.1016/j.immuni.2023.06.016.
McGeer EG, McGeer PL. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis. 2010;19(1):355–61. https://doi.org/10.3233/JAD-2010-1219.
Li Z, Wu H, Luo Y, Tan X. Correlation of serum complement factor 5a level with inflammatory response and cognitive function in patients with Alzheimer’s disease of different severity. BMC Neurol. 2023;23(1):319. https://doi.org/10.1186/s12883-023-03256-w.
Liu L, Liu X, Zhou J, Wu W, Li X. An improved graph Laplacian regularization method for identifying biomarkers of Alzheimer’s disease. J Theor Biol. 2022;543:111121. https://doi.org/10.1016/j.jtbi.2022.111121.
Barrett T, Suzek TO, Troup DB, Wilhite SE, Ngau WC, Ledoux P, et al. NCBI GEO: mining millions of expression profiles–database and tools. Nucleic Acids Res. 2005;33(Database issue):D562-6. https://doi.org/10.1093/nar/gki022.
Smyth GK. Limma: linear models for microarray data. Bioinformatics and computational biology solutions using R and Bioconductor. 2005. pp. 397–420.
Li D, Cao R, Dong W, Cheng M, Pan X, Hu Z, et al. Identification of potential biomarkers for ankylosing spondylitis based on bioinformatics analysis. BMC Musculoskelet Disord. 2023;24(1):413. https://doi.org/10.1186/s12891-023-06550-3.
Zhu J, Meng H, Zhang L, Li Y. Exploring the molecular mechanism of comorbidity of autism spectrum disorder and inflammatory bowel disease by combining multiple data sets. J Transl Med. 2023;21(1):372. https://doi.org/10.1186/s12967-023-04218-z.
Han L, Wang J, Ji XB, Wang ZY, Wang Y, Zhang LY, et al. Transcriptomics analysis identifies the presence of upregulated ribosomal housekeeping genes in the alveolar macrophages of patients with smoking-induced chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2021;16:2653–64. https://doi.org/10.2147/COPD.S313252.
Davis AP, Grondin CJ, Johnson RJ, Sciaky D, Wiegers J, Wiegers TC, et al. Comparative Toxicogenomics Database (CTD): update 2021. Nucleic Acids Res. 2021;49(D1):D1138–43. https://doi.org/10.1093/nar/gkaa891.
Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, et al. GeneCards Version 3: the human gene integrator. Database (Oxford). 2010;2010:baq020. https://doi.org/10.1093/database/baq020.
Zhang Z, Zheng Y, Bian X, Wang M, Chou J, Liu H, et al. Identification of key genes and pathways associated with oxidative stress in periodontitis. Oxid Med Cell Longev. 2022;2022:9728172. https://doi.org/10.1155/2022/9728172.
Qu S, Liu S, Qiu W, Liu J, Wang H. Screening of autophagy genes as prognostic indicators for glioma patients. Am J Transl Res. 2020;12(9):5320–31.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. https://doi.org/10.1186/1471-2105-9-559.
von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31(1):258–61. https://doi.org/10.1093/nar/gkg034.
Dunn PJ, Thomson E, Campbell J, Smith T, Desborough V, Wiser J, et al. ImmPort: shared research data for bioinformatics and immunology. In: 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). IEEE; 2015. pp. 607–10.
Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. https://doi.org/10.1186/1471-2105-14-7.
Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17(1):218. https://doi.org/10.1186/s13059-016-1070-5.
Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40. https://doi.org/10.1093/bioinformatics/btr260.
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–3. https://doi.org/10.1093/bioinformatics/btp101.
Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303. https://doi.org/10.1007/978-1-60761-987-1_18.
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168–74. https://doi.org/10.1001/archpsyc.63.2.168.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30. https://doi.org/10.1038/s41588-019-0358-2.
Bellenguez C, Kucukali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54(4):412–36. https://doi.org/10.1038/s41588-022-01024-z.
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–9. https://doi.org/10.1038/ng.439.
Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49(9):1373–84. https://doi.org/10.1038/ng.3916.
Chatterjee P, Pedrini S, Ashton NJ, Tegg M, Goozee K, Singh AK, et al. Diagnostic and prognostic plasma biomarkers for preclinical Alzheimer’s disease. Alzheimers Dement. 2022;18(6):1141–54. https://doi.org/10.1002/alz.12447.
Benedet AL, Mila-Aloma M, Vrillon A, Ashton NJ, Pascoal TA, Lussier F, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78(12):1471–83. https://doi.org/10.1001/jamaneurol.2021.3671.
Shi H, Koronyo Y, Rentsendorj A, Regis GC, Sheyn J, Fuchs DT, et al. Identification of early pericyte loss and vascular amyloidosis in Alzheimer’s disease retina. Acta Neuropathol. 2020;139(5):813–36. https://doi.org/10.1007/s00401-020-02134-w.
Miners JS, Schulz I, Love S. Differing associations between Abeta accumulation, hypoperfusion, blood-brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J Cereb Blood Flow Metab. 2018;38(1):103–15. https://doi.org/10.1177/0271678X17690761.
Wang J, Fan DY, Li HY, He CY, Shen YY, Zeng GH, et al. Dynamic changes of CSF sPDGFRbeta during ageing and AD progression and associations with CSF ATN biomarkers. Mol Neurodegener. 2022;17(1):9. https://doi.org/10.1186/s13024-021-00512-w.
Li X, Long J, He T, Belshaw R, Scott J. Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer’s disease. Sci Rep. 2015;5:12393. https://doi.org/10.1038/srep12393.
Pillai JA, Bebek G, Khrestian M, Bena J, Bergmann CC, Bush WS, et al. TNFRSF1B gene variants and related soluble TNFR2 levels impact resilience in Alzheimer’s disease. Front Aging Neurosci. 2021;13:638922. https://doi.org/10.3389/fnagi.2021.638922.
Brai E, Alina Raio N, Alberi L. Notch1 hallmarks fibrillary depositions in sporadic Alzheimer’s disease. Acta Neuropathol Commun. 2016;4(1):64. https://doi.org/10.1186/s40478-016-0327-2.
Baron BW, Pytel P. Expression pattern of the BCL6 and ITM2B proteins in normal human brains and in Alzheimer disease. Appl Immunohistochem Mol Morphol. 2017;25(7):489–96. https://doi.org/10.1097/PAI.0000000000000329.
Mancuso R, Fryatt G, Cleal M, Obst J, Pipi E, Monzon-Sandoval J, et al. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain. 2019;142(10):3243–64. https://doi.org/10.1093/brain/awz241.
Folch J, Patraca I, Martinez N, Pedros I, Petrov D, Ettcheto M, et al. The role of leptin in the sporadic form of Alzheimer’s disease. Interactions with the adipokines amylin, ghrelin and the pituitary hormone prolactin. Life Sci. 2015;140:19–28. https://doi.org/10.1016/j.lfs.2015.05.002.
Lin E, Kuo PH, Liu YL, Yang AC, Tsai SJ. Polymorphisms of the DNA repair gene EXO1 modulate cognitive aging in old adults in a Taiwanese population. DNA Repair (Amst). 2019;78:1–6. https://doi.org/10.1016/j.dnarep.2019.03.013.
Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, Olsen ML. The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. 2016;132(1):1–21. https://doi.org/10.1007/s00401-016-1553-1.
Szego EM, Csorba A, Janaky T, Kekesi KA, Abraham IM, Morotz GM, et al. Effects of estrogen on beta-amyloid-induced cholinergic cell death in the nucleus basalis magnocellularis. Neuroendocrinology. 2011;93(2):90–105. https://doi.org/10.1159/000321119.
Soles-Tarres I, Cabezas-Llobet N, Vaudry D, Xifro X. Protective effects of pituitary adenylate cyclase-activating polypeptide and vasoactive intestinal peptide against cognitive decline in neurodegenerative diseases. Front Cell Neurosci. 2020;14:221. https://doi.org/10.3389/fncel.2020.00221.
Wen YF, Xiao XW, Zhou L, Jiang YL, Zhu Y, Guo LN, et al. Mutations in GBA, SNCA, and VPS35 are not associated with Alzheimer’s disease in a Chinese population: a case-control study. Neural Regen Res. 2022;17(3):682–9. https://doi.org/10.4103/1673-5374.321000.
Katayama T, Sawada J, Takahashi K, Yahara O, Hasebe N. Meta-analysis of cerebrospinal fluid neuron-specific enolase levels in Alzheimer’s disease, Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Alzheimers Res Ther. 2021;13(1):163. https://doi.org/10.1186/s13195-021-00907-3.
Lau A, Bourkas M, Lu YQQ, Ostrowski LA, Weber-Adrian D, Figueiredo C, et al. Functional amyloids and their possible influence on Alzheimer disease. Discoveries (Craiova). 2017;5(4):e79. https://doi.org/10.15190/d.2017.9.
Bogdan S, Puscion-Jakubik A, Klimiuk K, Socha K, Kochanowicz J, Gorodkiewicz E. UCHL1 and proteasome in blood serum in relation to dietary habits, concentration of selected antioxidant minerals and total antioxidant status among patients with Alzheimer’s disease. J Clin Med. 2022;11(2):412. https://doi.org/10.3390/jcm11020412.
Nguyen KV. Encephalomalacia/gliosis, deep venous thrombosis, and cancer in Arg393His antithrombin Hanoi and the potential impact of the beta-amyloid precursor protein (APP) on thrombosis and cancer. AIMS Neurosci. 2022;9(2):175–215. https://doi.org/10.3934/Neuroscience.2022010.
Wu X, Shen Q, Chang H, Li J, Xing D. Promoted CD4(+) T cell-derived IFN-gamma/IL-10 by photobiomodulation therapy modulates neurogenesis to ameliorate cognitive deficits in APP/PS1 and 3xTg-AD mice. J Neuroinflammation. 2022;19(1):253. https://doi.org/10.1186/s12974-022-02617-5.
Wang Y, Wu X, Deng X, Ma Y, Huang S, Wang Y. Association of CD14-260 (-159) C/T and Alzheimer’s disease: systematic review and trial sequential analyses. J Neural Transm (Vienna). 2018;125(9):1313–8. https://doi.org/10.1007/s00702-018-1896-y.
Shu J, Li N, Wei W, Zhang L. Detection of molecular signatures and pathways shared by Alzheimer’s disease and type 2 diabetes. Gene. 2022;810:146070. https://doi.org/10.1016/j.gene.2021.146070.
Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Abeta plaques. Cell Rep. 2019;27(4):1293-306.e6. https://doi.org/10.1016/j.celrep.2019.03.099.
Costa AS, Guerini FR, Arosio B, Galimberti D, Zanzottera M, Bianchi A, et al. SNARE complex polymorphisms associate with alterations of visual selective attention in Alzheimer’s disease. J Alzheimers Dis. 2019;69(1):179–88. https://doi.org/10.3233/JAD-190147.
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–8. https://doi.org/10.1038/nature18629.
Markarian M, Krattli RP Jr, Baddour JD, Alikhani L, Giedzinski E, Usmani MT, et al. Glia-selective deletion of complement C1q prevents radiation-induced cognitive deficits and neuroinflammation. Cancer Res. 2021;81(7):1732–44. https://doi.org/10.1158/0008-5472.CAN-20-2565.
Xavier S, Sahu RK, Landes SG, Yu J, Taylor RP, Ayyadevara S, et al. Pericytes and immune cells contribute to complement activation in tubulointerstitial fibrosis. Am J Physiol Renal Physiol. 2017;312(3):F516–32. https://doi.org/10.1152/ajprenal.00604.2016.
Bai Y, Wang M, Zhao J, Bai H, Zhang X, Wang J, et al. Comparative transcriptome analysis reveals immunoregulation mechanism of lncRNA-mRNA in gill and skin of large yellow croaker (Larimichthys crocea) in response to Cryptocaryon irritans infection. BMC Genomics. 2022;23(1):206. https://doi.org/10.1186/s12864-022-08431-w.
Yang JX, Maria TC, Zhou B, Xiao FL, Wang M, Mao YJ, et al. Quercetin improves immune function in Arbor Acre broilers through activation of NF-kappaB signaling pathway. Poult Sci. 2020;99(4):2305. https://doi.org/10.1016/j.psj.2020.03.003.
Qin Y, Sun B, Zhang F, Wang Y, Shen B, Liu Y, et al. Sox7 is involved in antibody-dependent endothelial cell activation and renal allograft injury via the Jagged1-Notch1 pathway. Exp Cell Res. 2019;375(2):20–7. https://doi.org/10.1016/j.yexcr.2019.01.008.
Cumpelik A, Heja D, Hu Y, Varano G, Ordikhani F, Roberto MP, et al. Dynamic regulation of B cell complement signaling is integral to germinal center responses. Nat Immunol. 2021;22(6):757–68. https://doi.org/10.1038/s41590-021-00926-0.
Bastaert F, Kheir S, Saint-Criq V, Villeret B, Dang PM, El-Benna J, et al. Pseudomonas aeruginosa LasB subverts alveolar macrophage activity by interfering with bacterial killing through downregulation of innate immune defense, reactive oxygen species generation, and complement activation. Front Immunol. 2018;9:1675. https://doi.org/10.3389/fimmu.2018.01675.
Crowson AN, Magro CM, Usmani A, McNutt NS. Immunoglobulin A-associated lymphocytic vasculopathy: a clinicopathologic study of eight patients. J Cutan Pathol. 2002;29(10):596–601. https://doi.org/10.1034/j.1600-0560.2002.291005.x.
Volk T, Pannicke U, Reisli I, Bulashevska A, Ritter J, Bjorkman A, et al. DCLRE1C (ARTEMIS) mutations causing phenotypes ranging from atypical severe combined immunodeficiency to mere antibody deficiency. Hum Mol Genet. 2015;24(25):7361–72. https://doi.org/10.1093/hmg/ddv437.
Brickshawana A, Hinson SR, Romero MF, Lucchinetti CF, Guo Y, Buttmann M, et al. Investigation of the KIR4.1 potassium channel as a putative antigen in patients with multiple sclerosis: a comparative study. Lancet Neurol. 2014;13(8):795–806. https://doi.org/10.1016/S1474-4422(14)70141-3.
Riboldi E, Frascaroli G, Transidico P, Luini W, Bernasconi S, Mancini F, et al. Benzydamine inhibits monocyte migration and MAPK activation induced by chemotactic agonists. Br J Pharmacol. 2003;140(2):377–83. https://doi.org/10.1038/sj.bjp.0705428.
Muk T, Stensballe A, Dmytriyeva O, Brunse A, Jiang PP, Thymann T, et al. Differential brain and cerebrospinal fluid proteomic responses to acute prenatal endotoxin exposure. Mol Neurobiol. 2022;59(4):2204–18. https://doi.org/10.1007/s12035-022-02753-2.
Du T, Li G, Luo H, Pan Y, Xu Q, Ma K. Hippocampal alpha-synuclein mediates depressive-like behaviors. Brain Behav Immun. 2021;95:226–37. https://doi.org/10.1016/j.bbi.2021.03.020.
Leiherer A, Stoemmer K, Muendlein A, Saely CH, Kinz E, Brandtner EM, et al. Quercetin impacts expression of metabolism- and obesity-associated genes in SGBS adipocytes. Nutrients. 2016;8(5):282. https://doi.org/10.3390/nu8050282.
Chen ZM, Cui Q, Zhao B, Song R, Zhang X, Yoshie O. SST: spatial and semantic transformers for multi-label image recognition. IEEE Trans Image Process. 2022;31:2570–83. https://doi.org/10.1109/TIP.2022.3148867.
Melms A, Malcherek G, Gern U, Sommer N, Weissert R, Wietholter H, et al. Thymectomy and azathioprine have no effect on the phenotype of CD4 T lymphocyte subsets in myasthenia gravis. J Neurol Neurosurg Psychiatry. 1993;56(1):46–51. https://doi.org/10.1136/jnnp.56.1.46.
Mejia JE, Willmott A, Levy E, Earnshaw WC, Larin Z. Functional complementation of a genetic deficiency with human artificial chromosomes. Am J Hum Genet. 2001;69(2):315–26. https://doi.org/10.1086/321977.
Yoon YH, Hwang HJ, Sung HJ, Heo SH, Kim DS, Hong SH, et al. Upregulation of complement factor H by SOCS-1/3(-)STAT4 in lung cancer. Cancers (Basel). 2019;11(4):471. https://doi.org/10.3390/cancers11040471.
Thomas AM, Gerogianni A, McAdam MB, Floisand Y, Lau C, Espevik T, et al. Complement component C5 and TLR molecule CD14 mediate heme-induced thromboinflammation in human blood. J Immunol. 2019;203(6):1571–8. https://doi.org/10.4049/jimmunol.1900047.
Cai XY, Wang ZF, Ge SW, Xu G. Identification of hub genes and immune-related pathways for membranous nephropathy by bioinformatics analysis. Front Physiol. 2022;13:914382. https://doi.org/10.3389/fphys.2022.914382.
De Schepper S, Ge JZ, Crowley G, Ferreira LSS, Garceau D, Toomey CE, et al. Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer’s disease. Nat Neurosci. 2023. https://doi.org/10.1038/s41593-023-01257-z.
Ekinci IB, Hizal M, Emiralioglu N, Ozcelik U, Yalcin E, Dogru D, et al. Differentially expressed genes associated with disease severity in siblings with cystic fibrosis. Pediatr Pulmonol. 2021;56(5):910–20. https://doi.org/10.1002/ppul.25237.
Santonicola P, Germoglio M, d’Abbusco DS, Adamo A. Functional characterization of Caenorhabditis elegans cbs-2 gene during meiosis. Sci Rep. 2020;10(1):20913. https://doi.org/10.1038/s41598-020-78006-w.
Altuna-Azkargorta M, Mendioroz-Iriarte M. Blood biomarkers in Alzheimer’s disease. Neurologia (Engl Ed). 2021;36(9):704–10. https://doi.org/10.1016/j.nrleng.2018.03.006.
Zhang Q, Li J, Weng L. Identification and validation of aging-related genes in Alzheimer’s disease. Front Neurosci. 2022;16:905722. https://doi.org/10.3389/fnins.2022.905722.
Deng Y, Feng Y, Lv Z, He J, Chen X, Wang C, et al. Machine learning models identify ferroptosis-related genes as potential diagnostic biomarkers for Alzheimer’s disease. Front Aging Neurosci. 2022;14:994130. https://doi.org/10.3389/fnagi.2022.994130.
Yuan M, Feng Y, Zhao M, Xu T, Li L, Guo K, et al. Identification and verification of genes associated with hypoxia microenvironment in Alzheimer’s disease. Sci Rep. 2023;13(1):16252. https://doi.org/10.1038/s41598-023-43595-9.
Wang H, Zhang Y, Zheng C, Yang S, Chen X, Wang H, et al. A 3-gene-based diagnostic signature in Alzheimer’s disease. Eur Neurol. 2022;85(1):6–13. https://doi.org/10.1159/000518727.
Hu D, Mo X, Jihang L, Huang C, Xie H, Jin L. Novel diagnostic biomarkers of oxidative stress, immunological characterization and experimental validation in Alzheimer’s disease. Aging (Albany NY). 2023;15(19):10389–406. https://doi.org/10.18632/aging.205084.
Lin Y, Zhang L, Gao M, Tang Z, Cheng X, Li H, et al. miR-6076 targets BCL6 in SH-SY5Y cells to regulate amyloid-beta-induced neuronal damage. J Cell Mol Med. 2023. https://doi.org/10.1111/jcmm.17999.
Song M, Xiong JX, Wang YY, Tang J, Zhang B, Bai Y. VIP enhances phagocytosis of fibrillar beta-amyloid by microglia and attenuates amyloid deposition in the brain of APP/PS1 mice. PLoS One. 2012;7(2):e29790. https://doi.org/10.1371/journal.pone.0029790.
Lemercier P, Vergallo A, Lista S, Zetterberg H, Blennow K, Potier MC, et al. Association of plasma Abeta40/Abeta42 ratio and brain Abeta accumulation: testing a whole-brain PLS-VIP approach in individuals at risk of Alzheimer’s disease. Neurobiol Aging. 2021;107:57–69. https://doi.org/10.1016/j.neurobiolaging.2021.07.005.
Acknowledgements
Thanks to all personnel for their efforts.
Funding
This work was supported by grants from the National Natural Science Foundation of Jiangsu Province (No. BK20221201), the Wuxi Municipal Health and Family Planning Commission Fund (No. M202137), Top Talent Support Program for Young and Middle-aged People of Wuxi Health Committee (No.BJ2023040).
Author information
Authors and Affiliations
Contributions
Conception and design of the research: XCZ; acquisition of data: BFT, MZZ, JL, RL and TM; analysis and interpretation of data: BFT, HXL and JMT; Statistical analysis: TM; drafting the manuscript: XCZ; revision of manuscript for important intellectual content: XCZ. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The protocols of the present study were approved by the Institutional Review Board of the Affiliated Wuxi No.2 People’s Hospital, Nanjing Medical University.
Consent for publication
Not Applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
A total of 187 up-regulated genes and 280 down-regulated genes in Alzheimer’s disease. Supplementary Figure 1. Quality Control Preprocess of included data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, XC., Tang, BF., Zhu, MZ. et al. Analysis of complement system and its related factors in Alzheimer’s disease. BMC Neurol 23, 446 (2023). https://doi.org/10.1186/s12883-023-03503-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-023-03503-0