
Abstract
Background
In Egypt, the characterization of Neuromyelitis Optica Spectrum Disorder (NMOSD) is lacking.
Objectives
To determine the demographics, clinical features, aquaporin4 antibodies (AQP4-IgG) status, and neuroimaging of Egyptian NMOSD patients.
Methods
Retrospective analysis of 70 NMOSD patients’ records from the MS clinic, Kasr Alainy hospital, between January 2013 and June 2018.
Results
Patients’ mean age was 34.9 ± 9.2 years, and the mean at disease onset was 28.9 ± 10.5 years. Fifty-nine patients had an initial monosymptomatic presentation. AQP4-IgG was measured using either enzyme-linked immunosorbent assay (ELISA) (22 patients) or cell-based assay (CBA) (34 patients). Six and 29 patients had positive results, respectively (p < 0.001). 84% had typical NMOSD brain lesions. Longitudinally extensive myelitis was detected in 49 patients, and 9 had either short segments or normal cords. Treatment failure was higher in seropositive patients. Rituximab significantly reduced the annualized relapse rate (ARR) compared to Azathioprine with a percentage reduction of (76.47 ± 13.28) and (10.21 ± 96.07), respectively (p = 0.04). Age at disease onset was the only independent predictor for disability (p < 0.01).
Conclusion
Treatment failure was higher in seropositive patients. However, there was no difference in clinical or radiological parameters between seropositive and seronegative patients. Patients, who are polysymptomatic or with older age of onset, are predicted to have higher future disability regardless of the AQP4-IgG status.
Similar content being viewed by others


Background
Neuromyelitis Optica Spectrum Disorder (NMOSD) is a rare inflammatory central nervous system (CNS) disorder of autoimmune etiology [1].
Classic presentations of the disease result from lesions in areas that generally express high levels of aquaporin 4; this refers to the optic nerves, spinal cord, dorsal medulla, brain stem, thalamus, and hypothalamus. More than 50% of patients are expected to lose vision and their ability to walk independently within 5 years of disease onset [2].
Aquaporin 4 immunoglobulins G (AQP4- IgG) present in more than three-quarters of patients constitutes a sensitive and highly specific serum marker of NMOSD, distinguishing the disease from multiple sclerosis [3].
The NMOSD diagnostic criteria published in 2015 are considered one of the core diagnostic characteristics [4].
Unlike multiple sclerosis (MS), the prominent geographical heterogenicity is not yet proven for NMOSD, and it is still unclear whether the disease phenotypes and demographic features vary among different populations.
Significantly few published reports of NMOSD appeared from the middle east [5,6,7], and to date, much about the disease characteristics in Egypt (one of the most heavily populated countries in the middle east and north Africa region) is still not known.
The current study aimed to elucidate the demographics, clinical features, AQP4- IgG status, neuroimaging, and predictors of disability progression of Egyptian patients with NMOSD.
It is the first study in Egypt to assess the AQP4-IgG using the cell-based assay (CBA) and compare it with the traditional enzyme-linked immunosorbent assay (ELISA).
Methods
Study design and participants
Retrospective analysis of patients’ medical records with a working diagnosis of NMO/NMOSD attending the multiple sclerosis clinic, Kasr Alainy hospital, Cairo University, between January 2013 and June 2018 was done.
Data collection
Detailed clinical, laboratory, and radiological data were extracted by expert neurologists specialized in autoimmune and inflammatory neurological diseases.
Patients who were diagnosed before 2015 had their diagnosis revised according to the 2015 international panel for NMO Diagnosis (IPND) [4]. All patients fit into the revised criteria and were given the diagnosis of NMOSD. A total of 70 patients were enrolled in the study after experienced neurologists reviewed the patients’ data, and patients with suspected alternate diagnoses or missing data were excluded.
Baseline Expanded Disability Status Scale (EDSS) [8] was the one recorded in the patient’s first visit, and the follow-up was the last scored in the outpatient clinic visit within 2 months before data acquisition. The number of relapses for each patient was recorded, and the annualized relapse rate (ARR) was defined as the number of confirmed relapses per year (verified by a neurologist within 7 days after symptom onset) [9]. Treatment failure was considered if new CNS symptoms and signs that lasted longer than 24 h with or without new lesions on gadolinium-enhanced magnetic resonance imaging (MRI) occurred despite the use of immunotherapies [10].
Imaging
Cranial and spinal MRI with gadolinium was performed when clinically indicated with 1.5 Tesla scanners. MS-like lesions were defined as lesions fulfilling the Barkhofs criteria for multiple sclerosis, and NMOSD typical brain lesions were defined as peri ependymal lesions surrounding the ventricles and the aqueduct, extensive lesions involving corticospinal tracts, hemispheric tumefactive or cloud-like enhancing lesions [11, 12].
Aquaporin4-IgG testing
Testing for AQP4- IgG was done within 1 month following relapses using either ELISA or CBA by indirect immunofluorescence. Patients tested by CBA were also tested for anti-myelin oligodendrocyte glycoprotein antibodies (anti-MOG-Abs). Only two patients were positive for anti-MOG-Abs and were excluded from the study as well. Cerebrospinal fluid (CSF) analysis for immunoglobulins G (IgG) index and oligoclonal bands (OCB) detection was done in selected subjects who had initially doubtful diagnoses.
Ethical consideration
The study protocol was approved by the neurology department review board and followed the principles outlined in the declaration of Helsinki. Informed written consent was taken from all participants prior to the enrollment.
Statistical analysis
SPSS version 18.0 was used for data analysis. Mean ± standard deviation (SD) described quantitative variables and medians with range for data that did not follow normality. The number and percentages described qualitative data, and Chi-square tested proportion independence. For comparing the mean values of two independent groups and more than two independent groups, parametric and non-parametric t-test and one-way ANOVA were used. A paired t-test was used for comparing the means of 2 dependent groups. P-values were significant at 0.05. Kruskal Wallis test was used for comparing the percent ARR reduction. Multiple linear regression analysis was used to detect disability predictors, and multivariate analysis was used to describe independent variables that might determine the risk of having an EDSS of > 5.
Results
Demographics, clinical and paraclinical characteristics of the study population
Clinical characteristics
Our study population included 70 NMOSD patients (58 females and 12 males) with ages ranging from18 to 64 years.
The demographics, clinical and paraclinical characteristics of the study population are outlined in Table 1.
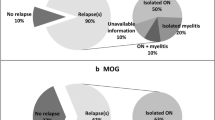
Twenty-seven (38.6%) patients presented with myelitis, 25 (35.7%) with optic neuritis, 6 (8.6%) with brainstem syndrome, 1 (1.4%) with area postrema syndrome, and 11 (15.7%) had a polysymptomatic presentation.
Motor and sensory symptoms were the most frequently reported by our patients throughout their disease course, followed by sphincteric, then visual complaints Table 2.
Aquaporin4-IgG status
Six out of 22 patients (37.5%) tested positive for AQP4-Ab by ELISA compared to 29/34 (85%) by CBA. (p < 0.001). Sixteen patients were negative by ELISA, and five were negative by CBA. Fourteen patients presented with typical clinical and imaging features fulfilling the 2015 consensus criteria for NMOSD with negative OCB and IgG index. These 14 patients were not tested for AQP4-IgG because they were on steroids or immunotherapies.
Immunotherapy
A total of 32 patients used Azathioprine, rituximab by 12, cyclophosphamide by 11, mitoxantrone by five, and mycophenolate mofetil by two over their disease course. The number of patients who have failed to adequately respond to those drugs when used as first, second, and third drugs is outlined in Table 3.
Comparing patients according to their AQP4-IgG status
Demographics and clinical characteristics
The clinical data and demographics of patients with positive and negative AQP4-IgG status are shown in Table 4.
Radiological findings
MS-like and non-specific MRI brain lesions were found in 6 aquaporin seropositive patients and 5 with a negative status. On the other hand, 29 seropositive and 30 patients with a negative status had NMOSD typical lesions. Table 5 shows MRI cervical and thoracic spine results concerning the aquaporin status.
Treatment response
Twenty-five patients have undergone plasma exchange for their acute relapses, one out of 15 with a positive aquaporin status, and 2 out of 8 with a negative status failed to respond to the procedure (p-value = 0.2). Response to immunotherapies concerning aquaporin status is shown in Table 6.
Comparing patients according to spinal cord lesions length
Forty-nine patients had long extensive transverse myelitis (LETM), and 9 had short segments or normal cords on either the cervical or the thoracic spine MRI scans; their mean ages and ages at disease onset were 34.7 ± 7.8, 35.3 ± .3 (p = 0.7), and 28.7 ± 9.2, 29.8 ± 6.5, (p = 0.6) respectively. No statistically significant difference was observed when comparing baseline or last assessed EDSS of both groups [5 ± 2.3, 3.9 ± 2, (p = 0.2) and 4.4 ± 2.4, 4 ± 2, (p = 0.7) respectively].
Annualized relapse rates with different immunotherapies
The percent reduction of ARR for rituximab and Azathioprine after excluding patients on treatment for less than one-year comparison yielded a statistical significance Table 7.
Regression analysis
Multiple linear regression analysis was done to detect disability predictors (Age at onset, disease duration, and the number of relapses). A significant regression equation was found (F (3,62) =4.25, p = 0.009) with R2 of 0.17, and the age at onset was the only independent predictor for disability (beta = 0.09, p < 0.01) Table 8.
Using multivariate analysis, the risk of having an EDSS score of more than 5 in patients with the polysymptomatic presentation was more than three times greater than initial monosymptomatic patients (OR = 3.8; 95% CI = 1.04–13.74; p = 0.04) Table 9.
Discussion
As far as we know, this is the first Egyptian study characterizing NMOSD to use CBA for diagnosis and to test for anti-MOG-Abs, besides having a larger number of patients than the previously reported in Egypt.
The mean age of disease onset in this study population was 28.9 ± 10.5 years, which was close to that reported by the only Egyptian study characterizing a cohort of 20 NMOSD patients (27.8 ± 13.71 years) [6], yet it remains lower than those outlined in other studies [7, 13,14,15].
In the current study, AQP4-IgG status was assessed in 22 patients using ELISA (the only assay available in Egypt before 2017), and in 34 patients using CBA, the latter assay conveys significantly higher sensitivity in detecting AQP4-IgG seropositivity. In this regard, we added the value of utilizing the CBA technique as the most sensitive assay currently available, recommended by the International consensus diagnostic criteria for neuromyelitis optica spectrum disorders and confirmed in previous studies [16, 17].
Despite using the most sensitive technique, AQP4-IgG is not detected in 10–40% of patients diagnosed with NMOSD [18]. Eighty-five percent of the patients tested by CBA in our study were positive for AQP4-IgG. This percentage is higher than in other cohorts from the United States, Malaysia, Thai, Japan, and Korea [19,20,21,22].
Statistically significant differences between AQP4-IgG seropositive and seronegative patients regarding the demographics, presenting symptoms, EDSS scores, MRI findings, and CSF results were not observed. It is crucial to note that the different AQP4-IgG testing techniques have likely diluted the differences between the two groups based on ELISA’s presumed false-negative results.
In our cohort, 8.1% of cervical or thoracic MRI lesions in AQP4-IgG positive patients were short segments. This underscores the importance of AQP4-IgG testing in patients having overlapping NMOSD and MS symptoms with short lesions to avoid MS misdiagnosis and, consequently, the use of therapies that might aggravate NMOSD in an account of the non-appreciation of the potential of short transverse myelitis to occur in NMOSD.
Rituximab appeared to provide better disease stabilization than Azathioprine in NMOSD patients in earlier studies [10, 23, 24]. Because of the availability and lower cost of Azathioprine compared to its counterparts, It is more frequently prescribed in our country.
In our patients, treatment with Azathioprine and rituximab resulted in a comparable reduction in ARR. However, after excluding patients on treatment for less than 1 year, rituximab showed significantly higher ARR percent reduction compared to Azathioprine.
Within the AQP4-IgG seropositive patients, the number of patients who failed immunotherapies was significantly higher than those with a negative status in our cohort. A study in 2018 described similar findings in their study that seropositive patients utilized significantly higher immunotherapies [25].
Disability determinants of our cohort revealed that age at onset was the only independent predictor by multivariant regression analysis; this is consistent with a study’s findings in 2012, which concluded that older age at disease onset was predictive of motor disability [26].
Some caveats need to be noted in our study, including the retrospective nature of the analysis, which could have introduced bias in data collection, the heterogenicity of AQP4-IgG testing techniques, and none of the patients tested using ELISA were tested for anti-MOG-Abs. The strengths included the relatively large number of patients enrolled, the use of CBA for diagnosis (first Egyptian NMO study to use the technique), and the anti-MOG-Ab testing in most patients.
Conclusion
Our findings contribute in a way to the emerging picture of NMOSD in the region. Treatment failure was higher in seropositive patients. However, there was no difference in clinical or radiological parameters between seropositive and seronegative patients. Patients, who are polysymptomatic or with older age of onset, are predicted to have higher future disability regardless of the AQP4-IgG status. Rituximab treatment resulted in a significant percentage reduction in ARR compared to Azathioprine.
Availability of data and materials
The datasets used and/or analyzed during the current study are available in the supplementary document.
Abbreviations
- NMOSD:
-
Neuromyelitis Optica Spectrum Disorder
- CNS:
-
Central nervous system
- AQP4-IgG:
-
Aquaporin 4 immunoglobulins G
- MS:
-
Multiple sclerosis
- ELISA:
-
Enzyme-linked immunosorbent assay
- CBA:
-
Cell-based assay
- IPND:
-
International Panel for NMO Diagnosis
- EDSS:
-
Expanded Disability Status Scale
- ARR:
-
Annualized relapse rate
- MRI:
-
Magnetic resonance imaging
- Anti-MOG-Abs:
-
Anti-myelin oligodendrocyte glycoprotein antibodies
- CSF:
-
Cerebrospinal fluid
- IgG:
-
Immunoglobulins G
- OCB:
-
Oligoclonal bands
- SD:
-
Standard deviation
- IQR:
-
Interquartile range
References
Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6(Suppl 9):805–15.
Zarei S, Eggert J, Franqui-Dominguez L, Carl Y, Boria F, Stukova M, et al. A comprehensive review of neuromyelitis optica and clinical characteristics of neuromyelitis optica patients in Puerto Rico. Surg Neurol Int. 2018;9:242.
Waters P, Jarius S, Littleton E, Leite MI, Jacob S, Gray B, et al. Aquaporin-4 antibodies in Neuromyelitis Optica and longitudinally extensive transverse myelitis. JAMA Neurol. 2008;65(Suppl 7):913–9.
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, International Panel for NMO Diagnosis, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(Suppl 2):177–89.
Holroyd KB, Aziz F, Szolics M, Alsaadi T, Levy M, Schiess N. Prevalence and characteristics of transverse myelitis and neuromyelitis optica spectrum disorders in the United Arab Emirates: a multicenter, retrospective study. Clin Exp Neuroimmunol. 2018;9(Suppl 3):155–61.
Salama S, Marouf H, Ihab Reda M, Mansour AR, Elkholy O, Levy M. Clinical and radiological characteristics of neuromyelitis optica spectrum disorder in the North Egyptian Nile Delta. J Neuroimmunol. 2018;324:22–5.
Shosha E, Al Asmi A, Nasim E, Inshasi J, Abdulla F, Al Malik Y, et al. Neuromyelitis optica spectrum disorders in Arabian gulf (NMOAG); establishment and initial characterization of a patient registry. Multiple Sclerosis Relat Disord. 2019;38:101448.
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(Suppl 11):1444–52.
Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(Suppl 5):387–401.
Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: a multicenter study of treatment efficacy. JAMA Neurol. 2014;71(Suppl 3):324–30.
Barkhof F, Filippi M, Miller DH, Scheltens P, Campi A, Polman CH, et al. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain. 1997;120(Suppl 11):2059–69.
Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol. 2006;63(Suppl 7):964–8.
Asgari N, Lillevang ST, Skejoe HP, Falah M, Stenager E, Kyvik KO. A population-based study of neuromyelitis optica in Caucasians. Neurology. 2011;76(Suppl 18):1589–95.
Jacob A, Panicker J, Lythgoe D, Elsone L, Mutch K, Wilson M, et al. The epidemiology of neuromyelitis optica amongst adults in the Merseyside county of United Kingdom. J Neurol. 2013;260(Suppl 8):2134–7.
Etemadifar M, Dashti M, Vosoughi R, Abtahi SH, Ramagopalan SV, Nasr Z. An epidemiological study of neuromyelitis optica in Isfahan. Mult Scler J. 2014;20(Suppl 14):1920–2.
Marignier R, Bernard-Valnet R, Giraudon P, Collongues N, Papeix C, Zephir H, et al. Aquaporin-4 antibody-negative neuromyelitis optica: distinct assay sensitivity-dependent entity. Neurology. 2013;80(Suppl 24):2194–200.
Sato DK, Nakashima I, Takahashi T, Misu T, Waters P, Kuroda H, et al. Aquaporin-4 antibody-positive cases beyond current diagnostic criteria for NMO spectrum disorders. Neurology. 2013;80(Suppl 24):2210–6.
Weinshenker BG, Wingerchuk DM. Neuromyelitis spectrum disorders. Mayo Clin Proc. 2017;92(Suppl 4):663–79.
Kim W, Park MS, Lee SH, Kim SH, Jung IJ, Takahashi T, et al. Characteristic brain magnetic resonance imaging abnormalities in central nervous system aquaporin-4 autoimmunity. Mult Scler. 2010;16(Suppl 10):1229–36.
Siritho S, Nakashima I, Takahashi T, Fujihara K, Prayoonwiwat N. AQP4 antibody-positive Thai cases: clinical features and diagnostic problems. Neurology. 2011;77(Suppl 9):827–34.
Mealy MA, Wingerchuk DM, Greenberg BM, Levy M. Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol. 2012;69(Suppl 9):1176–80.
Abdullah S, Wong WF, Tan CT. The prevalence of anti-aquaporin 4 antibody in patients with idiopathic inflammatory demyelinating diseases presented to a tertiary hospital in malaysia: presentation and prognosis. Mult Scler Int. 2017;6:1359761. https://doi.org/10.1155/2017/1359761.
Jeong IH, Park B, Kim SH, Hyun JW, Joo J, Kim HJ. Comparative analysis of treatment outcomes in patients with neuromyelitis optica spectrum disorder using multifaceted endpoints. Mult Scler. 2016;22(Suppl 3):329–39.
Stellmann JP, Krumbholz M, Friede T, Gahlen A, Borisow N, Fischer K, et al. Immunotherapies in neuromyelitis optica spectrum disorder: efficacy and predictors of response. J Neurol Neurosurg Psychiatry. 2017;88(Suppl 8):639–47.
Kunadison S, Tungkasereerak C, Saetang S, Mekawichai P. Comparison of clinical features between aquaporin-4 antibody seropositive and seronegative patients in neuromyelitis optica and neuromyelitis optica spectrum disorder. Neurol Asia. 2018;23(Suppl 1):55–9.
Kitley J, Leite MI, Nakashima I, Waters P, McNeillis B, Brown R, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain. 2012;135(Suppl 6):1834–49.
Acknowledgments
The authors would like to express their gratitude to the patients for their participation and cooperation in this study.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
NAK was the idea founder, shared in the patient collection, and the supervisor in all the steps. WA was responsible for the laboratory work. NMS shared in the patient collection. HSS shared in the patient collection. AH shared in the patient collection. MIH shared in the patient collection. NTA shared in the patient collection. DA shared in the patient collection. SMS was responsible for the laboratory work. SSA shared in the patient collection. NT shared in the patient collection. AMF shared in the patient collection and did the data analysis. AE shared in the patient collection and in writing the manuscript. AHR wrote and revised the manuscript, shared in the analysis of the results, and is the submitting and corresponding author. All authors read and approved the final manuscript.
Authors’ information
NAK is a professor of neurology at Cairo University.
WA is a lecturer of clinical pathology at Cairo University.
NMS is a professor of neurology at Cairo University.
HSS is a professor of neurology at Cairo University.
AH is a professor of neurology at Cairo University.
MIH is an assistant professor of neurology at Cairo University.
NTA is a professor of neurology at Cairo University.
DA is a lecturer of neurology at Cairo University.
SMS is a professor of clinical pathology at Cairo University.
SSA is an assistant lecturer of neurology at Cairo University.
NT is a lecturer of internal medicine at Cairo University.
AMF is a lecturer of neurology at Cairo University.
AE is an assistant professor of neurology at Cairo University.
AHR is a lecturer of neurology at Cairo University.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The neurology department, Cairo University review board, approved the study protocol, which followed the principles outlined in Helsinki’s declaration. Informed written consent was taken from all participants before the enrollment to participate in the study.
Consent for publication
Informed written consent for publishing identifying information in an online open-access publication was taken from all participants before the enrollment.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kishk, N.A., Abdelfattah, W., Shalaby, N.M. et al. The aquaporin4-IgG status and how it affects the clinical features and treatment response in NMOSD patients in Egypt. BMC Neurol 21, 53 (2021). https://doi.org/10.1186/s12883-021-02083-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-021-02083-1