
Abstract
Background
We encountered two unrelated individuals suffering from neurological disorders, including epilepsy and scoliosis.
Case presentation
Whole-exome sequencing identified the same recurrent, de novo, pathogenic variant in NUS1 [NM_138459.4:c.691 + 1C > A] in both individuals. This variant is located in the conserved cis-prenyltransferase domain of the nuclear undecaprenyl pyrophosphate synthase 1 gene (NUS1), which encodes the Nogo-B receptor, an essential catalyst for protein glycosylation. This variant was confirmed to create a new splice donor site, resulting in aberrant RNA splicing resulting in a 91-bp deletion in exon 3 in both individuals. The mutant mRNA was partially degraded by nonsense mediated mRNA decay. To date, only four de novo variants and one homozygous variant have been reported in NUS1, which cause developmental and epileptic encephalopathy, early onset Parkinson’s disease, and a congenital disorder of glycosylation. Seven patients, including our two patients, have presented with epileptic seizures and intellectual disabilities.
Conclusions
Our study strongly supports the finding that this recurrent, de novo, variant in NUS1 causes developmental and epileptic encephalopathy with involuntary movement, ataxia and scoliosis.
Similar content being viewed by others


Background
The NUS1 (nuclear undecaprenyl pyrophosphate synthase 1) gene encodes the Nogo-B receptor (NgBR) [1, 2], which interacts with dehydrodolichyl diphosphate synthase complex subunit (DHDDS) and promotes cis-prenyltransferase (cis-PTase) activity. NgBR is an essential catalyst of the dolichol monophosphate (Dol-P) biosynthetic machinery in eukaryotic cells [3, 4]. The well-conserved C-terminus domain of cis-PTase in NgBR has intrinsic effects for protein structure stabilization, in association with N-glycans. To date, four de novo NUS1 variants have been reported in association with developmental and epileptic encephalopathy (DEE) and early-onset Parkinson’s disease, and one homozygous NUS1 variant has been associated with a congenital disorder of glycosylation. Interestingly, two de novo variants [5] and a pair of compound heterozygous variants [6] in DHDDS have been reported in five patients with DEE seizures or congenital glycosylation defects, suggesting that pathogenic variants in the NgBR–DHDDS pathway may cause neurological disorders. Here, we report two unrelated Japanese patients with a novel, recurrent, de novo NUS1 variant, who presented with epileptic seizures with involuntary movement, ataxia, intellectual disability and scoliosis.
Case presentation
The patient 1 was the second child born to non-consanguineous, healthy parents. Her elder brother had febrile seizures during childhood. She was born spontaneously, at full term, with no asphyxia. Her birth weight was 2826 g (− 0.44 SD). She gained head control at 4 months of age and sat without support at 7 months of age. She experienced febrile seizures at 9 months of age and generalized tonic-clonic convulsions without fever at 14 months of age, at which time valproic acid (VPA) was administered. Tremulous myoclonus of the extremities was also observed. She walked without support at 20 months of age, spoke a meaningful word at 10 months of age and two-word phrases at 24 months of age. Her developmental quotient was 77 at 2 years of age. Her seizures occurred once per year until the age of 6 years and 5 months; however, an increase in the VPA dosage lessened an episode of convulsive attack, after which her seizures disappeared. Her electroencephalograms (EEG) showed 3-Hz, diffuse, spike-and-slow-wave complexes with a 7-Hz slow wave background at 8 years of age (Additional file 1: Figure S1A), which became worsened at 15 years of age. However, the treatment with VPA and LEV significantly lessened 3-Hz diffuse spike-and-wave complexes and only 3-Hz high-amplitude slow wave bursts were infrequently recorded during sleep at 17 years of age (Additional file 1: Figure S1B). Her brain magnetic resonance imaging (MRI) results were normal at 6 years of age (Additional file 1: Figure S1C, S1D) and at 15 years of age (Additional file 1: Figure S1E, S1F). At the age of 17 years, her height was 157.4 cm (− 0.12 SD) and her weight was 41.9 kg (− 1.42 SD). She had no dysmorphic features except for scoliosis which needed a surgical correction at 15 years of age. She showed dysgraphia, due to tremulous myoclonus of the bilateral extremities (Additional file 4: Movie S1). She showed no behavioral disorders, such as autistic spectrum disorders or attention deficit/hyperkinetic disorder.
Additional file 4: Movie S1. Involuntary movements of Patient 1 at 17 years of age. Patient 1 shows frequent eye blinking, multifocal facial twitching, tremulous myoclonus of the upper extremities, mild dysmetria, and clumsiness of diadochokinesis.
In patient 1, trio (sequencing with parents) WES was performed, and 5 de novo variants were detected (Additional file 5: Table S1 and Additional file 6: Table S2 and Additional file 7: Supplemental method). Patient 1 had a splice site variant in NUS1 [NM_138459.4:c.691 + 1C > A] on chromosome 6q22.1 (chr6: 118,015,344). This variant was absent from public databases (allele frequency was 0 in ExAC, gnomAD, ESP6500, HGVD, ToMMo and in-house 575 Japanese exome controls). Multiple in-silico evaluation scores for predicting the pathogenicity of DNA sequence alternations suggest that this variant is deleterious: Mutation Taster (http://www.mutationtaster.org/) returned a value of disease causing; CADD (https://cadd.gs.washington.edu/) returned a value of 25.9; and Fathmm (http://fathmm.biocompute.org.uk/) returned a value of deleterious. This splice variant is predicted [7] to create a new splice donor site, which could change the reading frame and introduce a premature termination codon (PTC).
The patient 2 was born normally to non-consanguineous, healthy parents. His birth weight was 3550 g. At the age of 6, it was noticed he had speech delay, clumsiness of the hands, and involuntary movements of the hands when he used chopsticks. At the age of 8 years, he was suspected to have a cerebellar atrophy, with seizures. Occasionally, jerky movements of the limbs also appeared. From 14 years of age, he gradually developed a gait abnormality. At the age of 37 years, he was admitted to hospital. On physical examination, he showed flat foot and limb ataxia. Clonazepam was remarkably effective for treating his gait disturbance. Laboratory examinations were all normal, including lactic acid, pyruvic acid, vitamins, the thyroid gland, ceruloplasmin, copper, lipoproteins, amino acid analysis and leukocyte lysosome enzyme activities (α-galactosidase, β-galactosidase, β-hexosaminidase and arylsulfatase). Genetic testing for dentatorubral-pallidoluysian atrophy was negative. At the age of 40 years, myoclonic jerks of the limbs developed, in addition to ataxia. He was diagnosed as progressive myoclonus epilepsy with an unknown cause. At the age of 42 years, scoliosis became apparent. Starting at the age of 48 years, he began to require assistance with walking. At the latest examination (59 years), he showed intellectual disability (equivalent to that of a 6-year-old), excessive blinking due to tenseness, and profound action myoclonus of the limbs, which could be referred to “hyperkinésie volitionnelle”. His eye pursuit was saccadic, and his speech was explosive. Tendon reflexes were slightly increased, and no sensory disturbances were observed. An EEG analyzing jerk-locked back averaging potentials suggested that the myoclonus emerged from the cortex. Examinations of MRI, nerve conduction studies, conventional EEGs, and laboratory examinations of the cerebrospinal fluid and blood were almost within normal ranges (Additional file 2: Figure S2A, S2B). Increasing the dosage of clonazepam up to 12 mg (0.5 mg × 24 tablets/day) did not alleviate neurological symptoms; however, the oral administration of 50 mg baclofen remarkably lessened myoclonus and slightly improved gait disturbance.
We performed proband-only WES in patient 2 and detected a splicing site alteration variant in NUS1 [c.691 + 1C > A] and a stop-gain variant in SPTAN1 [c.2311G > T:p.(Glu771*)]. These two variants were confirmed by Sanger sequencing using the parents’ DNA, and only the NUS1 variant [c.691 + 1G > A] occurred de novo (Fig. 1a and Additional file 7: Supplemental method). SPTAN1 variant was inherited from healthy mother. Therefore, its pathogenicity should be minimal in the patient.

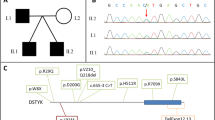
a Schematic presentation of the familial pedigrees of patients 1 and 2, with electropherograms of the heterozygous NUS1 variant occurring de novo. b Schematic presentation of the NUS1 gene structure, with identified variants that result in abnormal cDNA. Based on the cDNA sequencing, the variant [c.691 + 1G > A] creates a new splice donor site in the middle of exon 3 [c.601_602], resulting in the loss of a 91-bp section of the NUS1 exon 3. c Comparison of the gene expression levels between a control and a patient. Patient LCLs were cultured either with no treatment (NT) or with cycloheximide (CHX) to test NMD involvement. Gene expression was normalized to that of GAPDH. Error bars represent the 10th to 90th percentiles. The vertical numbers (Y-axis) represent the levels of relative gene expression. d Pathogenic NUS1 variants (including the current variant) mapped to the gene and the protein. A prenyltransferase domain is encoded by the middle of exons 2 to 5. Wild-type protein has 293 amino acids, and the prenyltransferase domain is composed of amino acids 156 to 292. This figure was designed using SMART software (http://smart.embl-heidelberg.de/)
To test whether this variant causes aberrant splicing, we examined the cDNA from both individuals’ lymphoblastoid cell lines, which revealed that the mutant allele has a 1-base alternation in the splice donor site (chr6:118,015,344) creating a new splice donor site of GT in exon 3 (chr6:118,015,253), resulting in a 91-bp deletion in the NUS1 exon 3 (Fig. 1b). Electropherograms of both individuals’ cDNA showed that a 91-bp region of exon 3 is missing. TA-cloning of the short RT-PCR product confirmed the same event in the mutant allele (Fig. 1b, Additional file 3: Figure S3A and Additional file 7: Supplemental method). This variant creates a new reading frame [c.601_691del:p.(Arg202Glnfs*9)] and produces a PTC at chr6:118,024,795 (73-bp upstream of the 3′ exon-exon junction) (Fig. 1b). According to the major rule of nonsense-mediated mRNA decay (NMD) [8], the mRNA of the mutant allele should be subjected to NMD. However, the results of both individuals’ cDNA sequencing chromatograms showed that cycloheximide (an NMD inhibitor) treatment did not recover the peak height of the electropherogram, suggesting that NMD is not involved. Thus, we performed a quantitative analysis using RT-PCR (see Additional file 7: Supplemental method). RT-PCR showed that the relative gene expression levels of NUS1 was slightly reduced in the patients’ LCLs, with possible minor recovery following cycloheximide treatment (Fig. 1c). These results support the prediction that the transcribed mRNA of the mutant allele is only partially subjected to NMD.
Discussion and conclusions
As the pLI score [9] of NUS1 in the ExAC browser is 0.87, suggesting that an intolerance to NUS1 loss-of-function variants, NUS1 variants may lead to diseases through haploinsufficiency. Diseases-associated, autosomal-dominant loss-of-function variants in NUS1 have been identified in patients with the following disorders: developmental delays and epileptic encephalopathy, [c.743delA:p.(Asp248Alafs*4)], [c.128_141dup:p.(Val48Profs*7)] and an approximately 1.3-kb deletion of exon 2 [5]; and a congenital disorder of glycosylation, [c.869G > A:p.(Arg290His)] [10]; and early-onset Parkinson’s disease, [c.691 + 3dupA] [11]. In addition, microdeletions involving NUS1 have been linked to pediatric epilepsy [12, 13]. The four loss-of-function variants identified in NUS1 occurred de novo (Table 1, Fig. 1d). These variants are located on the C-terminus cis-PTase domain, which is well-conserved in mammals. The cis-PTase domain interacts with DHDDS, and NgBR influences N-linked protein glycosylation via the regulation of cis-PTase activity [3] (Fig. 1d). A previous study [10] reported a homozygous variant, [c.869G > A:p.(Arg290His)] (Fig. 1d), in siblings with a congenital glycosylation disorder, congenital scoliosis and developmental delay, and refractory epilepsy. This mutation was found to decrease cis-PTase activity in the patients’ fibroblasts and caused a defect in the dolichol biosynthesis pathway. There are some phenotypic similarities among patients with NUS1 variants, regardless of inheritance patterns. The two individuals with the de novo [c.691 + 1C > A] variant identified in our study share similar phenotypes: epilepsy, involuntary movement, ataxia, intellectual disability, and scoliosis (Table 1).
The newly identified variant in this study created a PTC 73 nucleotides upstream of the last exon-exon junction (Fig. 1b), which should be subjected to NMD according to the < 50-bp rule of escaping from NMD [8]. We speculate that NMD was not fully induced in the studied patients, based on the results of our comparative gene expression analyses (Fig. 1c). A previous study [11] of the splice site mutation [c.691 + 3dupA] showed a larger decrease in mRNA expression compared with the results in our study. This inconsistency may be due to differences in the variants or to different cells being tested. Although the mutant allele is expressed, our study predicted that the variant results in aberrant splicing, resulting in the expressed NgBR lacking an important functional domain.
In conclusion, we found a recurrent, de novo variant in NUS1, [c.691 + 1C > A], in two unrelated individuals. Both individuals had a similar phenotype: epil7epsy, involuntary movement, ataxia, intellectual disability, and scoliosis. This study strongly suggests that loss-of-function variants in NUS1 that result in the loss of the cis-PTase domain in the C-terminus of NgBR may cause neurological disorders with scoliosis.
Availability of data and materials
The datasets generated and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
Abbreviations
- cis-PTase:
-
cis-prenyltransferase
- DEE:
-
Developmental and epileptic encephalopathy
- DMSO:
-
Dimethyl sulfoxide
- EEG:
-
Electroencephalogram
- LCL:
-
Lymphoblastoid cell lines
- MRI:
-
Magnetic resonance imaging
- NgBR:
-
Nogo-B receptor
- NMD:
-
Nonsense mediated mRNA decay
- NUS1:
-
Nuclear undecaprenyl pyrophosphate synthase 1
- PTC:
-
Premature termination codon
- SNV:
-
Single nucleotide variant
- VPA:
-
Valproic acid
- WES:
-
Whole-exome sequencing
References
Grabinska KA, Edani BH, Park EJ, Kraehling JR, Sessa WC. A conserved C-terminal RXG motif in the NgBR subunit of cis-prenyltransferase is critical for prenyltransferase activity. J Biol Chem. 2017;292(42):17351–61.
Miao RQ, Gao Y, Harrison KD, Prendergast J, Acevedo LM, Yu J, et al. Identification of a receptor necessary for Nogo-B stimulated chemotaxis and morphogenesis of endothelial cells. Proc Natl Acad Sci U S A. 2006;103(29):10997–1002.
Harrison KD, Park EJ, Gao N, Kuo A, Rush JS, Waechter CJ, et al. Nogo-B receptor is necessary for cellular dolichol biosynthesis and protein N-glycosylation. EMBO J. 2011;30(12):2490–500.
Shridas P, Rush JS, Waechter CJ. Identification and characterization of a cDNA encoding a long-chain cis-isoprenyltranferase involved in dolichyl monophosphate biosynthesis in the ER of brain cells. Biochem Biophys Res Commun. 2003;312(4):1349–56.
Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am J Hum Genet. 2017;101(5):664–85.
Sabry S, Vuillaumier-Barrot S, Mintet E, Fasseu M, Valayannopoulos V, Heron D, et al. A case of fatal type I congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity. Orphanet J Rare Dis. 2016;11(1):84.
Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4(3):311–23.
Lindeboom RG, Supek F, Lehner B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat Genet. 2016;48(10):1112–8.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
Park EJ, Grabinska KA, Guan Z, Stranecky V, Hartmannova H, Hodanova K, et al. Mutation of Nogo-B receptor, a subunit of cis-prenyltransferase, causes a congenital disorder of glycosylation. Cell Metab. 2014;20(3):448–57.
Guo JF, Zhang L, Li K, Mei JP, Xue J, Chen J, et al. Coding mutations in NUS1 contribute to Parkinson's disease. Proc Natl Acad Sci U S A. 2018;115(45):11567–72.
Milani D, Cagnoli GA, Baccarin M, Alfei E, Guerneri S, Esposito S. Insights into 6q21-q22: Refinement of the critical region for acro-cardio-facial syndrome. Congenit Anom. 2016;56(4):187–9.
Szafranski P, Von Allmen GK, Graham BH, Wilfong AA, Kang SH, Ferreira JA, et al. 6q22.1 microdeletion and susceptibility to pediatric epilepsy. Eur J Hum Genet. 2015;23(2):173–9.
Acknowledgements
We thank all subjects for participating in this study. We thank Lisa Giles, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Ethical approval and consent to participate
The Institutional Review Boards of Yokohama City University Graduate School of Medicine and Showa University School of Medicine approved the study protocol. Written informed consent was obtained from parents of patients, in accordance with the Japanese regulatory requirements.
Funding
This work was supported by AMED, under grant numbers JP18ek0109280, JP18dm0107090, JP18ek0109301, JP18ek0109348 and JP18kk020500; JSPS KAKENHI, under grant numbers JP17H01539, JP17K16132, JP16H05357, JP16H06254, JP17K10080, and JP17K15630; and grants from the Ministry of Health, Labour and Welfare, the Takeda Science Foundation, and The Ichiro Kanehara Foundation for the Promotion of Medical Science & Medical Care. The role of the funding body: Genetic analysis was supported by the fundings.
Author information
Authors and Affiliations
Contributions
Genetic analysis: KD, SM, TM, AT, NM, SM, and NM. Clinical evaluation: YK, KW, HD, FT, HO, and MK. Manuscript writing: KD, MK, HD, and SM. Manuscript supervision: NM. All authors read and approved the final version of the manuscript and ensure this in the case.
Corresponding author
Ethics declarations
Consent for publication
We obtained written informed consent for publication from all the patients and parents of case reports.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
(EEG and MRI of Patient 1). Interictal electroencephalogram (EEG) of Patient 1. A 3-s burst of 3-Hz, frontal-dominant, diffuse, spike-and-slow wave complexes is shown on an EEG at the age of 8 years (A). an EEG at 17 years (B) shows a similar burst of 3-Hz, high-amplitude (> 300 μV), slow waves, but no noticeable spike discharges. Brain MRIs for Patient 1 at the age of 6 years (C and D) and 15 years (E and F). T2-weighted axial images (C and E) and T1-weighted midsagittal images (D and F) show normal findings.
Additional file 2: Figure S2.
(MRI of Patient 2). Brain MRIs of Patient 2 at the age of 56 years (A and B). T2-weighted axial image (A) and T1- weighted midsagittal image (B) show normal findings.
Additional file 3: Figure S3.
A Agarose gel electrophoresis of cDNA fragments. B Gene expression levels of NUS1, normalized to those of actin. RT-PCR primers were same as those reported by Guo et al. [11]. Gene expression level was normalized to the expression level of actin: 5′-CCGGAAGATGGAAAAGCAGA-3′ (forward), 5′-TCCTTTCCTCCACAAGCCT-3′ (reverse). Gene expression levels were compared to those of control (ctrl) and no cycloheximide treatment (NT) conditions.
Additional file 5: Table S1.
Process of variant filtering for patient 1.
Additional file 6: Table S2.
Detailed information for the five de novo variants found in patient 1, including in silico prediction scores and allele frequencies.
Additional file 7:
Supplemental Method.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Den, K., Kudo, Y., Kato, M. et al. Recurrent NUS1 canonical splice donor site mutation in two unrelated individuals with epilepsy, myoclonus, ataxia and scoliosis - a case report. BMC Neurol 19, 253 (2019). https://doi.org/10.1186/s12883-019-1489-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-019-1489-x