
Abstract
Background
Anti-Myelin oligodendrocyte glycoprotein (MOG) antibodies are detected in various demyelinating diseases, such as pediatric acute disseminated encephalomyelitis (ADEM), recurrent optic neuritis, and aquaporin-4 antibody-seronegative neuromyelitis optica spectrum disorder. We present a patient who developed anti-MOG antibody-positive ADEM following infectious mononucleosis (IM) due to Epstein–Barr virus (EBV) infection.
Case presentation
A 36-year-old healthy man developed paresthesia of bilateral lower extremities and urinary retention 8 days after the onset of IM due to primary EBV infection. The MRI revealed the lesions in the cervical spinal cord, the conus medullaris, and the internal capsule. An examination of the cerebrospinal fluid revealed pleocytosis. Cell-based immunoassays revealed positivity for anti-MOG antibody with a titer of 1:1024 and negativity for anti-aquaporin-4 antibody. His symptoms quickly improved after steroid pulse therapy followed by oral betamethasone. Anti-MOG antibody titer at the 6-month follow-up was negative.
Conclusions
This case suggests that primary EBV infection would trigger anti-MOG antibody-positive ADEM. Adult ADEM patients can be positive for anti-MOG antibody, the titers of which correlate well with the neurological symptoms.
Similar content being viewed by others

Background
Myelin–oligodendrocyte glycoprotein (MOG) is exclusively expressed on the surface of oligodendrocytes in the central nervous system (CNS). Anti-MOG antibody is predominantly detected in pediatric acute disseminated encephalomyelitis (ADEM), recurrent optic neuritis, and aquaporin-4 antibody-seronegative neuromyelitis optica spectrum disorder (NMOSD). Recent studies proposed that anti-MOG antibody-associated demyelinating diseases were indeed a clinical spectrum in pediatric patients and that their clinical features were different from those of multiple sclerosis and NMOSD with anti-aquaporin-4 (AQP4) antibody [1, 2]. ADEM is a heterogeneous syndrome that is occasionally triggered by an antecedent infection [3]. A patient with anti-MOG antibody-positive longitudinally extensive transverse myelitis (LTEM) that developed after infection with influenza virus was previously reported [4]. However, no anti-MOG antibody-positive ADEM cases with a preceding viral infection other than influenza have been reported till date. Here we present a patient who developed anti-MOG antibody-positive ADEM following infectious mononucleosis (IM) due to primary Epstein–Barr virus (EBV) infection.
Case presentation
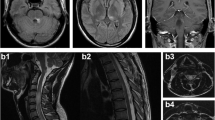
A 36-year-old healthy man developed fever and right cervical lymphadenopathy. Laboratory analysis showed elevated white blood count (10,390/mm3 with 33% neutrophil, 51% lymphocyte, and 12% atypical lymphocytes), elevated liver enzymes (aspartate transaminase, 193 U/l; alanine transaminase, 413 U/l). Serological studies indicated primary EBV infection (EBV viral capsid antigen [VCA] IgM, positive at 1:40; EBV VCA IgG, positive at 1:160, EBV nuclear antigen IgG, negative). Serologic testing for human immunodeficiency virus antibody was negative. Based on these clinical features, the patient was diagnosed with IM due to primary EBV infection. However, 8 days after onset, the patient developed paresthesia of bilateral lower extremities and urinary retention, which were exacerbated over the next few days. The patient was alert and oriented but had a high fever of 38.5 °C. Neurological examination revealed normal cranial nerves and no weakness in limbs; however, unstable gait with hyperreflexia, sensory disturbance in the entire area below the T7 level, and dysuria that required urethral catheterization were present. Laboratory analysis showed normal white blood count and decreasing liver enzyme levels. Antinuclear and SS-A antibody levels were within normal limits. Cerebrospinal fluid (CSF) examination showed pleocytosis (76/mm3), protein concentration of 104.3 mg/dl, IgG index of 0.61, the absence of oligoclonal IgG bands. In addition, IgG and IgM antibodies to EBV VCA and polymerase chain reaction for EBV DNA were negative in the CSF. These findings excluded the direct presence of EBV in the CNS. Additionally, polymerase chain reaction for herpes simplex virus 1, herpes simplex virus 2, and varicella-zoster virus DNA were negative in the CSF. IgG and IgM antibodies to cytomegalovirus were negative in the CSF. These findings excluded viral myelitis. Spinal MRI showed a T2-hyperintense lesion predominantly in the central gray matter extending from C2 to C6 (Fig. 1). Brain MRI showed a fluid-attenuated inversion recovery-hyperintense lesion in the left posterior limb of the internal capsule (Fig. 1). Nerve conduction studies of the left upper and lower extremities showed normal motor and sensory function. Cell-based immunoassays revealed positivity for anti-MOG antibody with a titer of 1:1024 and negativity for anti-AQP4 antibody [2]. Therefore, the patient was started on immunosuppressive therapy with intravenous methylprednisolone (IVMP) for 3 consecutive days, followed by oral betamethasone (2 mg/day). The gadolinium-enhanced spinal MRI after the start of therapy revealed slight gadolinium enhancement of the conus medullaris surface (Fig. 1). However, shortly after IVMP initiation, his symptoms demonstrated significant improvement, and urethral catheter was removed 9 days after the start of IVMP. His sensory disturbance and gait instability was completely resolved 2 weeks after IVMP initiation. Oral betamethasone was tapered following IVMP, and he was discharged without any symptoms or sequelae. Follow-up MRI 1 month after IVMP showed reduction in all CNS lesions. Anti-MOG antibody titer at the 6-month follow-up was negative. No symptomatic recurrence was observed during follow-up evaluation at 11 months after onset. Clinical course, the CSF and MRI findings, and the response to immunosuppressive therapy were most consistent with the diagnosis of anti-MOG antibody-positive ADEM [3, 5].

Spinal cord T2-weighted MRI shows a hyperintense lesion extending from C2 to C6 on the sagittal view (a), and predominantly in the central gray matter on the axial view at C4 (b. arrow) and C6 level (c. arrow). Gadolinium-enhanced T1-weighted MRI shows slight gadolinium enhancement within the conus medullaris surface (d. arrow heads). Brain fluid-attenuated inversion recovery MRI shows a hyperintense lesion in the left crus posterius capsulae internae (e. arrow)
Discussion and Conclusions
We present a case of a patient who developed anti-MOG antibody-positive ADEM following IM. In our patient, ADEM occurred relatively early i.e., 8 days after IM onset. However, the absence of EBV genome in the CSF samples is strong evidence for an autoimmune pathogenesis of neurological signs following IM. The present case illustrates two important clinical issues. First, adult ADEM patients can be positive for anti-MOG antibody, the titers of which correlated well with neurological symptoms. Among pediatric ADEM cases, pleocytosis, spinal cord lesions characterized by LTEM, and better outcomes were observed more frequently in patients with anti-MOG antibody than in those with negative titers. Anti-MOG antibody titers of monophasic ADEM patients declined or became negative over the course of months to years [6]. However, patients with persistently high anti-MOG antibody titers experienced a recurrent disease course [6, 7]. The anti-MOG antibody titer of the present case became negative and did not show recurrence. Thus, assessment for anti-MOG antibody titers in adult ADEM patients might be useful in predicting prognosis and determining the course of disease management.
Second, primary EBV infection triggers anti-MOG antibody-positive ADEM. Antecedent infections were reported in 46% of adult ADEM patients [3]. However, those were usually nonspecific upper respiratory tract infections, and systemic viral infections preceding ADEM were rarely reported in adult patients [3]. While several studies previously reported ADEM and LTEM cases associated with EBV infection [8–14], anti-MOG antibody titers were not examined in any of the studies. Recent reports proposed the presence of cross-reactivity between EBV and myelin proteins [15] and between MOG and EBV nuclear antigen [16]. Anti-MOG antibody was detected in 20% of patients with IM due to primary EBV infection without neurological manifestations [17]. These findings implicate EBV infection as a potential trigger for anti-MOG antibody production. However, a potential specific molecular mimicry between antibodies produced in response to EBV antigens and MOG remains unclear. The incidence of neurological involvement in IM was reported to range be 0.37–7.3% [8], and LTEM and ADEM remain very rare complications of EBV infection [8–10]. Therefore, we propose that anti-MOG antibody production might result from synergistic effects of additional unknown factors in response to EBV infection.
In conclusion, this case highlights the possibility that primary EBV infection triggers anti-MOG antibody-positive ADEM. Future studies are necessary to confirm the role of EBV in the pathogenesis of anti-MOG antibody-associated demyelinating diseases.
Abbreviations
- ADEM:
-
Acute disseminated encephalomyelitis
- AQP4:
-
Aquaporin-4
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- DNA:
-
Deoxyribonucleic acid
- EBV:
-
Epstein-Barr virus
- IM:
-
Infectious mononucleosis
- IVMP:
-
Intravenous methylprednisolone
- LTEM:
-
Longitudinally extensive transverse myelitis
- MOG:
-
Myelin oligodendrocyte glycoprotein
- MRI:
-
Magnetic resonance imaging
- NMOSD:
-
Neuromyelitis optica spectrum disorder
- VCA:
-
Viral capsid antigen
References
Reindl M, Di Pauli F, Rostásy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol. 2013;9:455–61.
Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM, Takahashi T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82:474–81.
Schwarz S, Mohr A, Knauth M, Wildemann B, Storch-Hagenlocher B. Acute disseminated encephalomyelitis: a follow-up study of 40 adult patients. Neurology. 2001;56:1313–8.
Amano H, Miyamoto N, Shimura H, Sato DK, Fujihara K, Ueno S, et al. Influenza-associated MOG antibody-positive longitudinally extensive transverse myelitis: a case report. BMC Neurol. 2014;14:224.
Tenembaum S, Chitnis T, Ness J, Hahn JS, International Pediatric MS Study Group. Acute disseminated encephalomyelitis. Neurology. 2007;68(16 Suppl 2):S23–36.
Baumann M, Sahin K, Lechner C, Hennes EM, Schanda K, Mader S, et al. Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J Neurol Neurosurg Psychiatry. 2015;86:265–72.
Baumann M, Hennes EM, Schanda K, Karenfort M, Kornek B, Seidl R, et al. Children with multiphasic disseminated encephalomyelitis and antibodies to the myelin oligodendrocyte glycoprotein (MOG): extending the spectrum of MOG antibody positive diseases. Mult Scler. 2016;22:1821–9.
Silverstein A, Steinberg G, Nathanson M. Nervous system involvement in infectious mononucleosis. The heralding and-or major manifestation. Arch Neurol. 1972;26:353–8.
Fujimoto H, Asaoka K, Imaizumi T, Ayabe M, Shoji H, Kaji M. Epstein-Barr virus infections of the central nervous system. Intern Med. 2003;42:33–40.
Tselis AC. Epstein-Barr virus infections of the nervous system. Handb Clin Neurol. 2014;123:285–305.
Mohsen H, Abu Zeinah GF, Elsotouhy AH, Mohamed K. Acute disseminated encephalomyelitis following infectious mononucleosis in a toddler. BMJ Case Rep 2013; doi:10.1136/bcr-2013-010048.
Caldas C, Bernicker E, Nogare AD, Luby JP. Case report: transverse myelitis associated with Epstein-Barr virus infection. Am J Med Sci. 1994;307:45–8.
Junker AK, Roland EH, Hahn G. Transverse myelitis and Epstein-Barr virus infection with delayed antibody responses. Neurology. 1991;41:1523–4.
Bahadori HR, Williams VC, Turner RP, Rumboldt Z, Reigart JR, Fowler SL, et al. Acute disseminated encephalomyelitis following infectious mononucleosis. J Child Neurol. 2007;22:324–8.
Lang HL, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol. 2002;3:940–3.
Wang H, Munger KL, Reindl M, O'Reilly EJ, Levin LI, Berger T, et al. Myelin oligodendrocyte glycoprotein antibodies and multiple sclerosis in healthy young adults. Neurology. 2008;71:1142–6.
Kakalacheva K, Regenass S, Wiesmayr S, Azzi T, Berger C, Dale RC, et al. Infectious mononucleosis triggers generation of IgG auto-antibodies against native myelin oligodendrocyte glycoprotein. Viruses. 2016;8:51.
Acknowledgments
Not applicable.
Funding
This work was supported by JSPS KAKENHI, Grant Number 15 K45678, from the Ministry of Education, Culture, Sports, Science and Technology, Japan. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The dataset supporting the conclusion of this article is included within the article.
Authors’ contributions
YN and HN examined and scripted the manuscript. HT, TH, SI, and FK helped to draft the manuscript and performed analyses. KK and TT performed anti-MOG antibody analysis. IN supported for the critical revision of the manuscript for intellectual content. All authors approved the contents of this case report.
Competing interest
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Ethics approval and consent to participate
The authors declare that ethics approval was not required for this case report.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nakamura, Y., Nakajima, H., Tani, H. et al. Anti-MOG antibody-positive ADEM following infectious mononucleosis due to a primary EBV infection: a case report. BMC Neurol 17, 76 (2017). https://doi.org/10.1186/s12883-017-0858-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-017-0858-6