
Abstract
Background
Kidney diseases with genetic etiology in children present with an overlapping spectrum of manifestations. We aimed to analyze the clinical utility of genetic testing in the diagnosis and management of suspected genetic kidney diseases in children.
Methods
In this retrospective study, children ≤ 18 years in whom a genetic test was ordered were included. Clinical indications for genetic testing were categorized as Glomerular diseases, nephrolithiasis and/or nephrocalcinoses, tubulopathies, cystic kidney diseases, congenital abnormality of kidney and urinary tract, chronic kidney disease of unknown aetiology and others. Clinical exome sequencing was the test of choice. Other genetic tests ordered were sanger sequencing, gene panel, multiplex ligation-dependent probe amplification and karyotyping. The pathogenicity of the genetic variant was interpreted as per the American College of Medical Genetics classification.
Results
A total of 86 samples were sent for genetic testing from 76 index children, 8 parents and 2 fetuses. A total of 74 variants were reported in 47 genes. Out of 74 variants, 42 were missense, 9 nonsense, 12 frameshifts, 1 indel, 5 affected the splicing regions and 5 were copy number variants. Thirty-two were homozygous, 36 heterozygous and 6 were hemizygous variants. Twenty-four children (31.6%) had pathogenic and 11 (14.5%) had likely pathogenic variants. Twenty-four children (31.6%) had variants of uncertain significance. No variants were reported in 17 children (22.3%). A genetic diagnosis was made in 35 children with an overall yield of 46%. The diagnostic yield was 29.4% for glomerular diseases, 53.8% for tubular disorders, 81% for nephrolithiasis and/or nephrocalcinoses, 60% for cystic kidney diseases and 50% for chronic kidney disease of unknown etiology. Genetic testing made a new diagnosis or changed the diagnosis in 15 children (19.7%).
Conclusion
Nearly half (46%) of the children tested for a genetic disease had a genetic diagnosis. Genetic testing confirmed the clinical diagnoses, changed the clinical diagnoses or made a new diagnosis which helped in personalized management.
Similar content being viewed by others


Introduction
With rapid advances in gene sequencing technology, an increasing number of Mendelian forms of kidney diseases are being identified in children with steroid-resistant nephrotic syndrome (SRNS), tubulopathy, nephrolithiasis and/or nephrocalcinosis, cystic kidney disease and hemolytic uremic syndrome. In some kidney diseases like congenital anomalies of the kidney and urinary tract (CAKUT), genetic etiology is not completely characterized. Many of the children with these kidney diseases progress to chronic kidney disease (CKD) [1, 2]. Nearly 30% of children and 5–30% of adults with CKD have one of the 450 genes mutated to explain the etiology of CKD [3]. Identifying monogenic causes has significant benefits in the management of children with kidney diseases. It ends the diagnostic journey by unequivocally establishing a diagnosis, allows informed treatment and helps in detecting and monitoring extra-renal complications. It also helps in detecting the disease in mildly symptomatic or asymptomatic family members by extended family screening. Knowledge of inheritance patterns and specific genetic variants guides further reproduction decisions and detection of the genetic variant in the fetus.
There is a significant shift in the paradigm of genetic testing in pediatric nephrology clinical practice with a move from a rare specialized and expensive test to the emergence as one of the common diagnostic methods used in the clinical setting. This is mainly due to the rapid progress in massively parallel sequencing and decreasing cost of massively parallel sequencing technology [4]. Massively parallel sequencing has also allowed the identification of new disease-causing genes and helped in understanding the molecular pathophysiology of many inherited diseases of childhood [3]. However, clear information regarding the likely outcome and impact of genetic testing is still missing. While the data on the clinical utility of genetic testing in specific disease groups in Indian children are available, information about its impact on diagnosis and management in an unselected group of children with kidney diseases is limited. We report our experience about the clinical utility of genetic testing in children with kidney diseases.
Methods
Study setting
The study was done at a tertiary care public nephrology teaching institute in India.
Ethical approval
The study was approved by institutional ethics committee registered at CDSCO with registration no ECR/143/Inst/GJ/2013/RR-19, reference letter: IKDRC-ITS EC/App/31Jul20/2. Informed consent was obtained from all subjects and/or their legal guardian(s).
Study design and Participants
This was a retrospective study where medical records of all children under the age of 18 years treated at a tertiary care nephrology center in whom a genetic etiology was suspected, and a genetic test was ordered between September 2016 to January 2021 were analyzed. Demographic details, history of consanguinity in parents, family history of similar illness, extrarenal and syndromic features were noted.
Disease categories
kidney diseases were categorized under the following disease categories: glomerular disease, nephrolithiasis and/or nephrocalcinosis, tubulopathy, cystic kidney disease, CKD with unknown etiology (CKDu), congenital abnormality of kidney and urinary tract (CAKUT) and others.
Genetic testing
The genetic testing method (karyotyping, multiplex ligation-dependent probe amplification (MLPA), Sanger sequencing, gene panel and clinical exome sequencing (CES)) was noted. The test results were interpreted as per the American College of Medical Genetics (ACMG) classification [5]. Where possible, family segregation studies were performed.
A genetic test was ordered by sending 5 ml blood in EDTA to a commercial laboratory.CES or targeted nephrotic syndrome panel were the investigations of choice when a genetic cause was suspected. Selective capture and sequencing of the protein-coding regions of the genome/genes were performed with a custom capture kit using the DNA extracted from blood. The libraries were sequenced to mean > 80-100X coverage on the Illumina sequencing platform (Illumina Inc., CA, USA). Genome Analysis Tool Kit best practices framework was followed for the identification of variants in the sample using Sentieon (v201808.01) [6]. Gene annotation of the variants was performed using the VEP program [7] against the Ensemble release 91 human gene model [8]. In addition to SNVs and small Indels, copy number variants (CNVs) were screened from sequence data using the ExomeDepth (v1.1.10) method [9]. Clinically relevant variants were annotated using published variants in literature and a set of diseases databases—ClinVar, OMIM, GWAS, HGMD (v2018.3) and SwissVar. Common variants were filtered based on allele frequency in 1000Genome Phase 3, ExAC (v1.0), gnomAD (v2.1), EVS, dbSNP (v151), 1000 Japanese Genome and internal Indian population database. The non-synonymous variant's effect was evaluated using multiple algorithms such as PolyPhen-2, SIFT, VariantTaster2 and LRT. Only non-synonymous and splice site variants found in the clinical exome panel consisting of 8332 genes were used for clinical interpretation. Silent variations that do not result in any change in amino acid in the coding region were not reported.
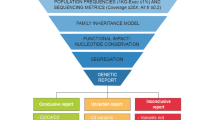
Sanger sequencing was done for some of the cases in parents of a proband when there was a strong suspicion of a particular gene, but more commonly to confirm the variant in the case of a variant of uncertain significance (VUS). MLPA was ordered for one of the selected case when there was no variant found in CES but when a copy number variation (CNV) was strongly suspected like in HUS. The SALSA MLPA Probemix P236 CFH Region from MRC Holland was used for the detection of deletions or duplications in the CFH, CFHR1, CFHR2, CFHR3, CFHR4 and CFHR5 genes in genomic DNA isolated from human peripheral whole blood specimens. Copy number differences of various exons between test and control DNA samples were detected by analyzing the MLPA peak patterns. Segregation analysis could not be done in all cases due to limited funds. The work flow diagram has been depicted in Fig. 1. The clinical impact of genetic testing was assessed by determining its utility in confirming the clinical diagnosis, making a new diagnosis, reclassifying a disease, reverse phenotyping, changing treatment and genetic counselling.

Flow Diagram to indicate the protocol followed for testing of samples
CES was done in 72 samples, targeted Nephrotic Syndrome gene panel in 2 (genes covered in Nephrotic Syndrome gene panels are given in Table 1), Sanger sequencing in 14 and MLPA in 1. Of the 14 Sanger sequencing performed, 2 were done to confirm CES variants, 8 in parents of children to confirm inheritance pattern and segregation, 2 for screening known genes in familial context or established syndrome, and 2 in the fetus. Karyotyping was done in 3 children.
Statistical analysis
Descriptive statistics were used for the study. The yield of genetic testing for various disease categories were expressed as percentage. The categorical variables were described as median. The numbers were small for any comparative statistical analysis.
Results
Patient characteristics
Genetic testing was performed in 86 individuals between September 2016 to January 2021, consisting of 76 index children, 8 parents, and 2 fetuses. Overall, the median age (IQR) of children at the onset of disease was 48 months (12–96 months). Forty out of 76 children (52.6%) were males. Nine families (10.5%) had a history of consanguinity. A family history of the same disease was present in only 4 families. The clinical disease categories are provided in Table 2. Glomerular diseases (44.7%) were the most common disease category for which a genetic test was ordered, in which the majority were steroid-resistant nephrotic syndrome (32.8%). Notably, 5.2% of the patients presented with kidney failure of unknown origin.
Variant identification and diagnostic yield
A total of 74 variants in 47 genes were reported in 59 out of 76 index children (77.6%). Seventeen children (22.3%) did not have any genetic variant. Out of all 74 variants 51 were novel and were not previously reported. Eighteen out of 51 novel variants were pathogenic/likely pathogenic and 33 were VUS. Forty-two variants were missense, 9 nonsense, 12 frameshifts, 1 indel, 5 variants affected the splicing regions and 5 were copy number variants (CNV).
A genetic diagnosis was made in 35 children (overall yield 46%; 35/76). Twenty-four children (31.5%) had pathogenic and 11(14.4%) had likely pathogenic variants. By disease category, the diagnostic yield was 29.4% in glomerular diseases (10 of 34 children), 53.8% in tubular disorders (7 of 13 children), 81% in nephrolithiasis and/or nephrocalcinosis (9 of 11 children), 60% in ciliopathies/cystic kidney diseases (6 of 10 children) and in 50% of CKD of unknown etiology (2 of 4 children) (Table 3). Of the 17 distinct monogenetic disorders detected, primary hyperoxaluria (n = 6) followed by Alport syndrome (n = 5) were the most common genetic diagnoses in the cohort. In 15 children (19.7%) genetic testing provided a new diagnosis or changed the clinical diagnosis. It revised the clinical diagnosis in 4 children. Clinical characteristics and details of genetic variants of children diagnosed with a genetic disease are depicted in Table 4. Twenty-four out of 76 children (31.5%) had variants of uncertain significance (VUS). Six children with pathogenic variants had also additional VUS. The details of children with VUS and children without a genetic variant are depicted in Tables 5 and 6 respectively.
The median age of the children with a genetic disease at disease onset was 24 months (range 10–108 months) compared to 48 months (range 48–82 months) in children without a genetic diagnosis. The majority, 28 (80%) had an autosomal recessive inheritance and 6 (17.1%) had a history of consanguinity. Syndromic features were noted in 6 children (17.1%).
Glomerular diseases
Among glomerular diseases, steroid-resistant nephrotic syndrome (SRNS) was the most common indication for ordering a genetic test (n = 25; 32.8%). All were initial SRNS. Seven children (28%) had a pathogenic variant, 9 (36%) had VUS while 9 children did not have any variant. Four of the seven (57%) with a pathogenic variant had the age of onset less than 24 months. Two children had a pathogenic variant in WT1 and 1 each in NPHS2, PLCE1, COL4A3, LAMB2 and NPHP1. One child with WT1 had clinical phenotype of Frasier vs Denys Drash syndrome (Case 37) and other with WT1 (Case 67) had SRNS without any other syndromic feature. Genetic testing resulted in a new diagnosis in 3 children (12%) with a clinical diagnosis of SRNS [nephronophthisis-1 (case 5), Pierson syndrome (case 24) and Frasier syndrome (case 67)]. In case 5 the patient was clinically diagnosed as FSGS as he had nephrotic syndrome at presentation along with kidney failure and biopsy had revealed sclerosed glomeruli. CES revealed nephronophthisis which gave the genetic diagnosis and predicted post kidney transplantation recurrence. In the other two children it helped in reverse phenotyping. In case 67 who was phenotypically female, karyotyping was done after CES which revealed 46 XY suggesting complete sex reversal. In case 24 with Pierson syndrome however there was no microcornea. In 5 children, immunosuppression was discontinued following genetic diagnosis. Nine children with VUS also had very severe disease resistant to both prednisolone and calcineurin inhibitors, similar to those with an identified genetic cause. Causality could not be established in those with VUS as genetic tests in parents were inconclusive and functional analysis could not be done. Four children with a pathogenic variant and 5 with VUS progressed to end-stage kidney disease (ESKD). In contrast, only 1 child without any genetic variant progressed to ESKD.
CES was done in all children with alternative complement pathway abnormality (atypical HUS n = 4, C3GN n = 2); however, MLPA was done in only 1 child due to cost constraints. VUS were identified in all children in WT1, SPTB, CFB, CFHR1 and CFHR3 genes. In 3 children with suspected Alport syndrome, diagnosis was confirmed in all (1 X linked Alport Syndrome and 2 Autosomal recessive Alport Syndrome).
Cystic kidney diseases
Ten children with suspected cystic kidney disease were evaluated for a genetic variant. Five had suspected nephronophthisis, 2 were suspected Bardet Biedl Syndrome, while 1 patient each was clinically diagnosed with glomerular cystic kidney disease, autosomal dominant polycystic kidney disease (ADPKD), autosomal recessive polycystic kidney disease (ARPKD). Five (50%) had extrarenal manifestations; retinitis pigmentosa in 3, polydactyly in 1and skeletal dysplasia in 1. Six (60%) had confirmed genetic disease out of which 4 had a pathogenic variant in SDCCAG8, DYNC2H1, NPHP 1, and CEP164 and 2 had likely pathogenic variant in PKHD1. Two had VUS in PKD1 and PKD2. In one child (case 1) the clinical diagnosis was reclassified from Bardet Biedl Syndrome to nephronophthisis 15. In Case 32, a new diagnosis of short-rib thoracic dysplasia was made. In one child (case 72) clinical diagnosis of ADPKD (large kidneys with cysts in kidneys and liver) was reclassified to ARPKD. One child (case 10) had no genetic variant despite having a strong phenotype of nephronophthisis realted ciliopathy (ESKD and retinitis pigmentosa with an affected sibling of a similar phenotype).
Nephrolithiases/nephrocalcinoses
Of 11 children with nephrolithiasis and/or nephrocalcinosis, clinical diagnoses were primary hyperoxaluria (n = 8), uric acid stone with hyperuricemia (n = 2) and familial hypomagnesemia, hypercalciuria and nephrocalcinosis (FHHNC) (n = 1). Nine (81%) had a pathogenic variant. Five had a pathogenic variant in the AGXT gene leading to the diagnosis of primary hyperoxaluria type 1. All of them had ESKD at presentation. One child (case 27) who had normal kidney function had a likely pathogenic variant in HOGA1. One child had a VUS in the GRHPR gene. Two (case 12, 14) with hyperuricemia, AKI and obstructive stones at 2 and 10 months of life were found to have a pathogenic variant in the HPRT1 gene leading to the diagnosis of Lesch Nyhan syndrome. Apart from hyperuricemia, there were no other features of Lesch Nyhan syndrome in these children. The child with suspected FHHNC had a VUS in Claudin 16.
Tubulopathies
Among 13 children with a clinical diagnosis of tubular disorders, 4 had Fanconi syndrome, 2 suspected distal RTA, 2 bartter syndrome, 2 rickets, 1 dent disease and 2 unknown tubulopathies. Seven (53.8%) had pathogenic variant; 1 SLC12A1, SLC4A1, SLCA2, FAH1, PHEX, CTNS and ATP6V0A4. No causal variants were identified in 3 children with unclear clinical diagnoses.
CKD with unknown etiologies
CES was done in four children with CKD with unknown etiology before transplant. One girl (case 35) had a Pathogenic variant in COL4A4 leading to the diagnosis of autosomal recessive Alport syndrome. On family screening, the younger sibling was found to harbor the same pathogenic variant leading to early diagnosis of Alport’s syndrome. One boy (case 44) had a homozygous pathogenic variant in COL4A5 while one child (case 64) had a homozygous VUS in the BBS4 gene. The other two children had no genetic variant.
Discussion
The study presents two points that are of interest. First, the feasibility of genetic testing in a clinical setting using a combination of methods for sequencing and second the impact of genetic diagnosis in the management of kidney disease and family screening in an unselected cohort. In the study, the genetic cause for kidney disease was identified in 35 out of 76 children (46%). The solve rate was high in children with nephrolithiasis and/or nephrocalcinosis (8/11;81%), ciliopathies/cystic kidney diseases (6/10; 60%), tubular disorders (7/13; 53.8%) and was least in glomerular diseases (10 of 34;29.4%). With the help of genetic testing, in 50% of children (2/4) with CKD with unknown etiology, a specific cause could be ascertained. Children with a pathogenic variant were younger at disease onset than those without a genetic etiology. This was similar to earlier reports, where the probability of having a pathogenic variant increase with younger age and decreases as age increases [10]. Although a majority of the children (81%) with genetic disease had a homozygous variant with autosomal recessive inheritance, family history was present in only 4. Hence it is important to note that the absence of family history should not be a factor in not suspecting a genetic cause [11].
The high yield in our cohort substantiates the use of genetic testing in establishing a molecular diagnosis. Low median age, 10% consanguinity and detailed phenotyping in the cohort before sending genetic test were probably predictive of the high diagnostic yield. In published studies, the diagnostic yield of genetic testing varies from 6.3 to 100% depending on the characteristics of the cohort and the method of analysis employed [3]. In a cohort of 127 patients ranging from newborn to 81 years, the overall solve rate of massively parallel sequencing by a kidney disease panel (KidneySeq v1, 177 genes) was 43% (54 of 127 patients) [10]. The solve rate was 46% in patients between 0–14 years, which decreased to 22% for patients > 30 years old. The solve rate (46%) in our cohort was similar. Though CES was not done in all the children with SRNS, the proportion (28%) of children with a pathogenic variant was similar to the median of 26% reported in earlier cohorts [3, 10, 12,13,14,15,16]. One-third of children had VUS, mostly novel with a phenotypically severe disease similar to those with a monogenic form, indicating a high probability of having a genetic cause.
Whole exome sequencing in patients < 25 years with either nephrolithiasis or nephrocalcinosis detected a monogenic cause in 29% in an earlier study [17]. However, 9 of 11 children (81%) with nephrolithiasis and nephrocalcinosis in the current study had a pathogenic variant, probably reflecting a carefully selected cohort where a genetic test was done. Similarly, the yield of a causal variant in tubulopathies in our cohort was high (53.8%) which was similar to the previous report in European cohorts [18, 19]. Two-thirds of children with cystic kidney disease had a pathogenic variant, which was much higher than reported in one study (12%) but similar to a median of 50% based on two earlier studies [3, 20,21,22].
CKDu is frequently seen in CKD cohorts; 6% at 12–15 years, 21% at 18–21 years and 36% of all cases with adult-onset CKD do not have a diagnosis [23]. Massively parallel sequencing has been increasingly used in the CKD population and is found to be useful in providing an alternative strategy to obtain a definitive diagnosis as recently demonstrated in 9 out of 92 patients by Lata et al. [24] and in 16 of 34 families (47%) in a cohort of 114 Irish families [25]. Two out of 4 children with CKDu (50%) had a pathogenic variant in our study. It established the diagnosis of autosomal recessive Alport syndrome and X linked Alport syndrome in these children.
In 20 out of 35 children with a genetic diagnosis, genetic testing correlated with the phenotype thereby confirming the diagnosis and further helped in prognostication, clinical management, and genetic counselling. For example, diagnosis of primary hyperoxaluria was confirmed in 6 children. Renal prognosis is different in different types of primary hyperoxaluria. Hence, genetic testing in hyperoxaluria not only helps establish the diagnosis but also to know the specific type of hyperoxaluria. In addition, it informs about the approach to transplant as Type I hyperoxaluria requires combined kidney-liver transplantation while in Type II, only kidney transplant would suffice.
In 15 children (19.7%) genetic testing provided a new diagnosis or revised the initial diagnosis. A correct diagnosis by genetic testing helps in counselling, facilitates living donor selection, and clarifies the risk of recurrence post-renal transplantation. For example, detection of a pathogenic variant in the NPHP1 gene established the diagnosis of nephronophthisis in a child suspected of FSGS progressed to ESKD (case 5). It not only unequivocally established the cause of kidney failure but also clarified disease recurrence risk post kidney transplantation. In case 72, the diagnosis was changed from ADPKD to ARPKD after the detection of a pathogenic variant in the PKHD1 gene. Reclassification or establishing a new diagnosis helps in reverse phenotyping in children. For example, in two children with CKD (case 36, 44) in whom etiology was unknown, diagnosis of Alport syndrome was made based on detection of homozygous pathogenic variant in COL4A4 and COL4A5 genes. This has important management implications as these children require screening for deafness as well as evaluation of the eyes. Similarly, detection of a heterozygous pathogenic variant in intron 9 of WT1 lead to the diagnosis of Frasier syndrome in a girl (case 67), who was then found to have 46 XY in karyotyping, complete sex reversal and gonadal dysgenesis. Further, immunosuppression was stopped as a therapeutic response was unlikely. This is a powerful demonstration of personalized medicine based on a genetic diagnosis.
Massively parallel sequencing has increasingly helped in identifying phenocopy. A child (case 70) with dense medullary nephrocalcinosis and suspected primary hyperoxaluria based on a high urine oxalate excretion, was diagnosed to have Bartter syndrome type 2 after detection of a homozygous pathogenic variant in the KCNJ 1 gene. It has important therapeutic as well as prognostic implications. The child did not have typical symptoms of Bartter syndrome. Repeat investigation following genetic results did reveal metabolic alkalosis and high urinary chloride establishing the correct diagnosis and helped in initiating appropriate treatment.
Diagnosis of genetic disease helps in the detection of disease in other family members as well as for antenatal counselling. For example, case36 was diagnosed with ARAS (pathogenic variant in COL4A4) when CES was done to evaluate the cause of CKD. Sanger sequencing of the same gene in her younger sibling who had microscopic hematuria, led to the discovery of the same variant (case 76). Detection of the same genetic variant in HPRT1 and AGXT gene in two fetuses as observed in index cases with Lesch nehan syndrome and hyperoxaluria respectively helped in appropriate counselling and termination of pregnancy.
While massively parallel sequencing based testing has many utilities, there are limitations too. Despite advances in bioinformatics, pathogenicity in some cases will remain uncertain. Twenty-four children (31.5%) had VUS, in whom definitive pathogenicity could not be established although phenotypically they were strongly suspected of having a genetic disease. Identification of VUS (mostly novel variants) as observed in a significant proportion of children in the current study poses challenges in interpretation and conveying the information to parents. In addition, segregation studies in the family to ascertain causality adds to the cost. A girl (case 11) with a heterozygous VUS in the PLCE1 gene developed recurrence of FSGS post kidney transplantation proving that the variant identified in the child was not causal. Hence, critical assessment of reported variants must occur before clinical decision-making is influenced by genetic findings. Besides VUS, 17 children did not have a genetic variant. Two siblings had a phenotype of Senior Loken syndrome and ESKD but no pathogenic variant was detected in one of the siblings (case 10) who was tested. Inability to detect causal variants could be due to limitations in testing strategies or identification of the variant in a gene that is yet to be associated with the phenotype or when the disease has a complex inheritance pattern (e.g. Digenic variants). Whole-exome sequencing could be useful in these cases as clinical exome sequencing might not detect a novel variant or copy number variations. These limitations and the requirement of additional testing in the family should be informed to parents before ordering genetic testing. In 6 children, a VUS was identified as a secondary variant along with a pathogenic variant. The significance of these variants could not be found out and they remain a clinical challenge. Also, being a retrospective review, detailed clinical information like GFR was lacking in many patients which is a major limitation of this study.
Conclusion
Genetic testing was useful in confirming a suspected diagnosis, making a new diagnosis, reverse phenotyping, genetic counselling, and personalized treatment in our cohort. Detailed phenotyping probably increased the yield of genetic testing. However, detection of a variant of uncertain significance remains a significant clinical challenge.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the NCBI repository at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA863410
References
Studies, N. A. P. R. T. a. C. NAPRTCS 2008 annual report. 2008. Retrieved from https://web.emmes.com/study/ped/annlrept/Annual%20Report%20-2008.pdf
Furth SL, Abraham AG, Jerry-Fluker J, Schwartz GJ, Benfield M, Kaskel F, et al. Metabolic abnormalities, cardiovascular disease risk factors, and GFR decline in children with chronic kidney disease. Clin J Am Soc Nephrol CJASN. 2011;6(9):2132–40. https://doi.org/10.2215/CJN.07100810.
Connaughton DM, Hildebrandt F. Personalized medicine in chronic kidney disease by detection of monogenic variants. Nephrol Dial Transplant. 2020;35(3):390–7. https://doi.org/10.1093/ndt/gfz028.
Behjati S, Tarpey PS. What is NGS? Arch Dis Child Educ Pract Ed. 2013;98(6):236–8. https://doi.org/10.1136/archdischild-2013-304340.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Kendig KI, Baheti S, Bockol MA, Drucker TM, Hart SN, Heldenbrand JR, et al. Sentieon DNASeq Variant Calling Workflow Demonstrates Strong Computational Performance and Accuracy. Front Genet. 2019;20(10):736. https://doi.org/10.3389/fgene.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–70. https://doi.org/10.1093/bioinformatics/btq330.
Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–61. https://doi.org/10.1093/nar/gkx1098.
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28(21):2747–54. https://doi.org/10.1093/bioinformatics/bts526.
Mansilla MA, Sompallae RR, Nishimura CJ, Kwitek AE, Kimble MJ, Freese ME, et al. Targeted broad-based genetic testing by next-generation sequencing informs diagnosis and facilitates management in patients with kidney diseases. Nephrol Dial Transplant. 2021;36(2):295–305. https://doi.org/10.1093/ndt/gfz173.
Hildebrandt F. Genetics of Kidney Diseases. Semin Nephrol. 2016;36(6):472–4. https://doi.org/10.1016/j.semnephrol.2016.09.011.
Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26(6):1279–89. https://doi.org/10.1681/ASN.2014050489.
Warejko JK, Tan W, Daga A, Schapiro D, Lawson JA, Shril S, et al. Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin J Am Soc Nephrol. 2018;13(1):53–62. https://doi.org/10.2215/CJN.04120417.
Trautmann A, Lipska-Ziętkiewicz BS, Schaefer F. Exploring the Clinical and Genetic Spectrum of Steroid Resistant Nephrotic Syndrome: The PodoNet Registry. Front Pediatr. 2018;17(6):200. https://doi.org/10.3389/fped.2018.00200.
Sampson MG, Gillies CE, Robertson CC, et al. Using population genetics to interrogate the monogenic nephrotic syndrome diagnosis in a case cohort. J Am Soc Nephrol. 2016;27:1970–83. https://doi.org/10.1681/ASN.2015050504.
Bierzynska A, McCarthy HJ, Soderquest K, et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. 2017;91:937–47. https://doi.org/10.1016/j.kint.2016.10.013.
Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int. 2018;93(1):204–13. https://doi.org/10.1016/j.kint.2017.06.025.
Bockenhauer D, Ashton E. Van’tHoff W, et al022 Genetic investigations in renal tubulopathies. Arch Dis Child. 2018;103:A9.
Ashton EJ, Legrand A, Benoit V, et al. Simultaneous sequencing of 37 genes identified causative variants in the majority of children with renal tubulopathies. Kidney Int. 2018;93:961–7. https://doi.org/10.1016/j.kint.2017.10.016.
Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identification of 99 novel variants in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet. 2013;132(8):865–84. https://doi.org/10.1007/s00439-013-1297-0.
Braun DA, Schueler M, Halbritter J, Gee HY, Porath JD, Lawson JA, et al. Whole exome sequencing identifies causative variants in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int. 2016;89(2):468–75. https://doi.org/10.1038/ki.2015.317.
Mallett AJ, McCarthy HJ, Ho G, et al. NGS and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney Int. 2017;92:1493–506. https://doi.org/10.1016/j.kint.2017.06.013.
Neild GH. Primary renal disease in young adults with renal failure. Nephrol Dial Transplant. 2010;25:1025–32. https://doi.org/10.1093/ndt/gfp653.
Lata S, Marasa M, Li YFD, et al. Whole-exome sequencing in adults with chronic kidney disease: a pilot study. Ann Intern Med. 2018;168:100–9. https://doi.org/10.7326/M17-1319.
Connaughton DM, Kennedy C, Shril S, et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019. https://doi.org/10.1016/j.kint.2018.10.031.
Acknowledgements
We acknowledge Dr Anil Vasudevan, Professor Pediatric Nephrology, St John’s Medical College, Bengaluru, India for his inputs in writing the manuscript.
Funding
Nil.
Author information
Authors and Affiliations
Contributions
AS conceptualised the study, collected data, analysed the data, wrote the manuscript and will act as guaranter of the article. SK collected the data and helped in writing the manuscript. KV collected data and helped in writing the manuscript. HP reviewed the manuscript and gave critical inputs.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Institute of Kidney Disease and Research Center-Institute of Transplantation Sciences Ethics committee registered at CDSCO with registration no ECR/143/Inst/GJ/2013/RR-19 (valid up to Nov 2024) approved the study. Reference letter: IKDRC-ITS EC/App/31Jul20/21. All methods were carried out in accordance with relevant guidelines and regulations.
Written informed consent were obtained from legal guardian(s).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Saha, A., Kapadia, S.F., Vala, K. et al. Clinical utility of genetic testing in Indian children with kidney diseases. BMC Nephrol 24, 212 (2023). https://doi.org/10.1186/s12882-023-03240-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-023-03240-z