
Abstract
Background
Hypocomplementemic urticarial vasculitis (HUV) is a rare systemic vasculitis. We aimed to describe the kidney involvement of HUV in a multicenter national cohort with an extended follow-up.
Methods
All patients with HUV (international Schwartz criteria) with a biopsy-proven kidney involvement, identified through a survey of the French Vasculitis Study Group (FVSG), were included. A systematic literature review on kidney involvement of HUV was performed.
Results
Twelve patients were included, among whom 8 had positive anti-C1q antibodies. All presented with proteinuria, from mild to nephrotic, and 8 displayed acute kidney injury (AKI), requiring temporary haemodialysis in 2. Kidney biopsy showed membrano-proliferative glomerulonephritis (MPGN) in 8 patients, pauci-immune crescentic GN or necrotizing vasculitis in 3 patients (with a mild to severe interstitial inflammation), and an isolated interstitial nephritis in 1 patient. C1q deposits were observed in the glomeruli (n = 6), tubules (n = 4) or renal arterioles (n = 3) of 8 patients. All patients received corticosteroids, and 9 were also treated with immunosuppressants or apheresis. After a mean follow-up of 8.9 years, 6 patients had a preserved renal function, but 2 patients had developed stage 3–4 chronic kidney disease (CKD) and 4 patients had reached end-stage kidney disease (ESKD), among whom 1 had received a kidney transplant.
Conclusion
Renal involvement of HUV can be responsible for severe AKI, CKD and ESRD. It is not always associated with circulating anti-C1q antibodies. Kidney biopsy shows mostly MPGN or crescentic GN, with frequent C1q deposits in the glomeruli, tubules or arterioles.
Similar content being viewed by others


Key messages
-
Kidney involvement in HUV can be life-threatening and lead to end-stage kidney disease.
-
Glomerular, vascular and tubulo-interstitial compartments can be injured, with frequent C1q deposits.
-
Systematic screening for kidney involvement during flares of HUV could accelerate diagnosis and treatment and reduce kidney damage.
Introduction
Hypocomplementemic urticarial vasculitis (HUV) is a rare systemic vasculitis, also called “McDuffie syndrome” or “anti-C1q vasculitis”. It was classified among immune complex small vessel vasculitides in the revised international Chapel Hill consensus conference in 2012 [1]. It was first described by McDuffie et al. in 1973 and then well defined in 1982 by Schwartz et al. [2, 3]. They proposed diagnostic criteria composed of 2 major criteria (recurrent urticarial lesions for at least 6 months and hypocomplementemia) associated with 2 minor criteria among the following: leukocytoclastic vasculitis (on cutaneous biopsy), arthralgia or arthritis, glomerulonephritis, ocular inflammation (uveitis or episcleritis), recurrent abdominal pain or anti-C1q antibody positivity. The frequency of kidney involvement associated with HUV ranges from 14 to 50% across different cohorts, and is poorly defined [4,5,6]. In the French national cohort published in 2015 by Jachiet et al. [5], a kidney involvement of HUV was observed in 10 (18%) patients, but detailed pathological lesions were not available. In addition, little is known on the long-term prognosis of kidney involvement in HUV, and no specific therapeutic strategy is defined in the literature. We describe here the clinical presentation, the pathological lesions and the therapeutic management of a multicentre cohort of 12 patients with biopsy-proven kidney involvement associated with HUV, identified through the French Vasculitis Study Group (FVSG) network.
Patients and methods
This is a multicentre retrospective study. All cases of patients with HUV, as defined by Schwartz criteria, and a biopsy-proven kidney involvement, identified through a survey sent to physicians from the French Study Group on Vasculitis (FSGV) network, were reviewed and included. Some of these patients were included in the national cohort by Jachiet et al. The following data was collected for each patient: demographic characteristics, systemic symptoms, skin lesions, adenopathy, rheumatic, ocular, eyes nose throat (ENT), lung, gastrointestinal, neurologic, cardiac involvement, and detailed renal parameters. AKI was defined according to the RIFLE criteria as R (risk), I (injury) and F (failure) [7]. Serum levels of C-reactive protein (CRP), complement fractions C3 and C4, anti-C1q antibodies, antinuclear antibodies (ANA), antiphospholipid antibodies, rheumatoid factor, cryoglobulinemia, anti-neutrophil cytoplasmic antibodies (ANCA) and protein electrophoresis were collected. All patients were tested for immunization against hepatitis B, C, HIV and syphilis. Results of skin biopsies were collected when available. All patients had at least one kidney biopsy, analysed by light microscopy and immunofluorescence. Electronic microscopy description was reported when available. The therapeutic strategies, global outcome and renal outcome were collected. Chronic kidney disease (CKD) was defined as a permanently altered kidney function with an estimated glomerular filtration rate (eGFR) < 60 mL/min/1.73m2 using the MDRD formula [8].
We then performed a systematic literature review on cases of HUV with renal involvement, using the following MESH terms: Hypocomplementemic urticarial vasculitis, Mac Duffie, kidney/renal involvement, acute kidney injury, chronic kidney disease, glomerulonephritis and kidney/renal biopsy.
For statistical analysis, continuous variables were compared using the Wilcoxon-Mann-Whitney test on mean or median values; categorical variables were compared using the Fisher’s exact test.
Results
Patients’ characteristics
Twelve patients with a biopsy-proven kidney involvement of HUV were identified, of whom 6 were previously included in the cohort of Jachiet et al. Their clinical and biological characteristics are detailed in Table 1. Two patients had a paediatric onset of HUV. Chronic urticarial lesions were present in all patients. Arthralgia were reported in 8 patients, with clinical arthritis in 3, and lymphadenopathies in 5 patients. Five patients displayed ocular involvement of HUV: anterior uveitis in 3 patients, conjunctivitis in 2, retinal involvement in two. Pulmonary symptoms (recurrent bronchitis) were described by 3 patients, among whom 1 had chronic respiratory failure. One patient displayed a pericarditis. Anti-C1q antibodies were positive in 8 patients. ANA were detected at some point during the follow-up in 7 patients, among whom one had anti-SSA and anti-double stranded DNA antibodies (patient #12), and another had anti-SSA antibodies and positive cryoglobulinemia (patient #7). ANCA antibodies were detected in 6 patients at some point during the follow-up, among whom 3 had anti-myeloperoxidase (MPO) antibodies, 1 anti-proteinase 3 (PR3) antibodies, and 2 no antigenic specificity. There was no difference in the characteristics of patients with or without ANCA, or with or without ANA, except for the higher frequency of ANA positivity in patients with ANCA, and of ANCA positivity in patients with ANA (Supplementary Tables 1 and 2).
Renal involvement of HUV
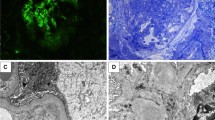
All patients presented with proteinuria, from mild to nephrotic. Eight patients presented with acute kidney injury (AKI), requiring temporary hemodialysis in 2 patients. One patient had stage R, 5 stage I and 3 stage F AKI. Pathological examination of kidney is detailed in Table 2 for the 12 patients, among whom 2 had a repeat biopsy. Kidney lesions were various, with different patterns of glomerular, vascular, and tubulo-interstitial involvements (Fig. 1). A membrano-proliferative GN (MPGN) was documented in 8 patients, with crescents in 7, and with a full-house immunofluorescence pattern in 1 patient. Four patients had active vascular lesions: 2 had a necrotizing vasculitis (patients #2 and #11), and 2 had immune deposits of IgG, IgM, C3 and C1q in vascular walls (patient #2 and #9). One patient (#12) had an isolated interstitial nephritis without glomerular or vascular changes. The repeat biopsy of patients #2 and #7 showed consistent glomerular and vascular patterns, with different levels of inflammatory and fibrotic lesions. Second biopsy of patient #9 showed severe chronic lesions and glomerulosclerosis. Electron microscopy analysis was performed in 2 patients. It showed, in patient #4, abundant and diffuse electron-dense segmental subepithelial and subendothelial granular deposits in capillary walls; in the second biopsy of patient #7, few glomerular electron-dense granular deposits.

Representative kidney pathological lesions observed in patients with Hypocomplementemic Urticarial Vasculitis from the present cohort. A Vasculitis with fibrinoid necrosis involving a pre-glomerular arteriole. Masson’s trichrome, × 200. B Global endocapillary proliferation with an interstitium containing a slight peri-venular inflammatory infiltrate. Masson’s trichrome, × 200. C Interstitial nephritis with mononuclear cells, oedema, and suffering tubules. Masson’s trichrome, × 200. D Abundant deposits of C1q within arteriole wall, capsule, and tubular basement membrane. Immunofluorescence, × 200
Treatment and outcome
Corticosteroids were prescribed in all patients, for all renal flares of HUV, as shown in Fig. 2A. Corticosteroids were tapered and continued for several years in some patients. An additional immunosuppressive therapy was prescribed in 9 patients. Rituximab was used in 5/12 patients (1 as a first line therapy, 3 after corticosteroids, 1 after cyclophosphamide). It allowed a remission in 4 patients (patient #9, with severe AKI initially requiring dialysis, was not in remission at last follow-up), and was reinfused successfully in 2 patients who relapsed. Patients were followed-up for an average of 8.9 years. The detailed evolution of patients is described in Fig. 3 and Supplementary Fig. 1. The total number of renal flares per patient ranged from 1 to 8. An infectious trigger for renal relapses was documented in 2 patients (one with pneumococcal pneumonia, and one with several relapses occurring after viral or bacterial infections). Plasma exchanges were used in 2 patients from this cohort: patient #2, for a relapse of HUV with severe AKI, which required also successively mycophenolate mofetil, cyclophosphamide and rituximab; and patient #9, for persistent severe AKI with massive proteinuria despite corticosteroids and rituximab. Patient #2 reached ESRD 2 years later, after an unplanned pregnancy, and received a kidney transplant without further relapse of HUV. Her second pregnancy was carried-out normally while she was transplanted, without HUV relapse.

Treatment of the kidney involvement of HUV (first, second and third line therapies). A In the 12 patients from the present cohort. B In the 26 patients from the literature

Treatment and renal outcome in 3 patients representative of the present cohort (patients #1, #2 and #10)
At last follow-up, all patients were alive. Patient #2 had received a kidney transplant, patients #3 and #7 were on chronic dialysis, patient #9 had stage 5 CKD, patients #10 had stage 3A CKD. The other 7 patients had a preserved eGFR (≥60 mL/min/1.73m2) without proteinuria. A severe complication of immunosuppressive therapy had occurred in 2 patients (one bacterial pneumonia and one cryptococcal meningitis).
We investigated the possible relationship between histological damage and HUV outcome, and the possible predictive factors for prognosis (Supplementary Tables 3, 4 and 5). Only baseline serum creatinine was predictive of CKD (eGFR < 60 mL/min/1.73m2) and of relapses.
Kidney involvement of HUV in the literature
Case reports of kidney involvement of HUV from the literature were collected and compared to patients from the present cohort (Table 3) [9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31]. The clinical presentation of the 26 patients with kidney involvement of HUV from the literature was consistent with the present work but high blood pressure was more frequent at diagnostic in our cohort (p = 0.005). The broad range of kidney lesions observed in the present cohort was also observed in the literature. Cryoglobulinemia and ANCAs antibodies were more frequent in our cohort (respectively p = 0.002 and 0.02) and the follow-up was longer (p = 0.002). Out of these 26 patients, 5 died, 4 reached ESRD and 2 developed stage 3 CKD.
Treatments administered to the patients from the literature are detailed in Fig. 2B. Two patients with kidney involvement received rituximab as a third line therapy: the first patient experienced a full recovery of kidney function after 12 months of dialysis dependence [31], the second was a child without available data on outcome [24]. Plasma exchanges were used in 2 patients from this cohort but were not sufficient and required additional treatment with rituximab or cyclophosphamide. Only 2 patients from the literature were treated with plasma exchanges for AKI (1 reached ESKD, the other reached remission) [13, 22].
Considering both the present cohort and patients from the literature (total n = 38), the presence of anti-C1q antibodies was not associated with outcomes: 41% of patients with positive anti-C1q had an unfavourable outcome (ESKD or death) versus 33% of patients without antiC1q (p = 0.74).
Discussion
This is the first study describing specifically biopsy-proven kidney involvement of HUV, with detailed pathological data and long term follow-up. We show that HUV can be associated with different glomerular lesions (mainly MPGN, but also crescentic GN, with or without immune glomerular deposits), lesions of renal arterioles (both acute and chronic, sometimes with C1q and immune complex deposits), and tubulo-interstitial inflammation (sometimes with tubular C1q and immune complex deposits). Notably, at the time of kidney biopsy, the interstitial fibrosis was often absent or mild, suggesting a rapid and recent occurrence of renal lesions. This kidney involvement can be life-threatening, with AKI requiring emergency dialysis, and can lead to ESKD. Yet, even with a severe initial presentation, a complete renal recovery can be obtained with corticosteroids, and possibly immunosuppressive therapy, in a majority of patients.
HUV is an immune-complex mediated systemic vasculitis affecting small vessels [1]. Immune complex deposits in vessel walls are usually documented on skin biopsies of urticarial lesions, and we show here that these deposits, comprising C1q, can also be encountered in renal arterioles.
Yet, immune deposits are not systematic in the kidney of patients with HUV, especially in glomeruli, where pauci-immune forms of crescentic GN can be encountered. An overlap between HUV and ANCA-associated vasculitis (AAV) could be discussed in these patients. Here, among the 6 patients with positive ANCA antibodies, only 4 had specific ANCA, among whom 3 displayed MPGN (classically not encountered in AAV) and only 1 displayed pauci-immune CGN. This last patient, indeed, both responded to HUV classification criteria (chronic urticaria, complement consumption, arthralgia, glomerulonephritis, and anti-C1q antibodies) and displayed features of AAV (arthralgia, glomerulonephritis and anti-MPO antibodies).
Similarly, the possible overlap between HUV and systemic lupus erythematosus (SLE) can be discussed in patients with positive ANA. Among the 7 patients with positive ANA, only 1 had anti-dsDNA, and displayed isolated interstitial nephritis. Only 1 patient with positive ANA displayed a MPGN with full-house immune deposits. Apart from this kidney involvement, this patient had no typical feature of SLE but displayed: biopsy-proven cutaneous leucocytoclastic vasculitis, angioedema, and recurrent bronchitis with chronic respiratory failure.
HUV is a rare disease, but its exact incidence is unknown [6]. Kidney involvement of HUV is even more rare: in the largest retrospective cohort of HUV, comprising 57 cases gathered in France over 20 years, 10 (18%) patients displayed kidney involvement of HUV [5]. In an American cohort comprising 18 patients with HUV, 9 (50%) patients displayed kidney involvement [4]. More recently, a Swedish cohort of 16 patients reported 5 patients with kidney involvement, among whom 2 reached ESKD [6]. MPGN, membranous GN, minimal change disease, focal glomerulopathy and anti-glomerular basement membrane nephritis have been described in the literature [32]. We confirm in this work the heterogeneity of clinical presentation and kidney pathological lesions in patients with HUV.
AntiC1q antibodies are IgG antibodies directed against the collagen-like region of C1q. They are encountered in HUV and in other diseases associated with immune complex deposition, such as mixed connective tissue disorder, SLE and post-infectious vasculitis [5]. The presence of anti-C1q antibodies has been associated with kidney involvement in SLE, and more specifically with crescentic lesions [33]. The presence of anti-C1q antibodies was only a minor criterion in Schwartz et al. [2], but was considered more discriminant in the revised international Chapel Hill consensus conference of 2012 [1]. However, the specificity and sensitivity of anti-C1q antibodies are unknown. Their presence is neither sufficient nor mandatory to diagnose HUV: 67% of patients from the present cohort, and 38% of patients from the literature had positive anti-C1q antibodies. In the French retrospective cohort by Jachiet et al., 55% of HUV patients had positive anti-C1q antibodies and 90% had a low C1q, and 88% from the American cohort by Wisnieski et al. had positive anti-C1q antibodies. The prognostic value of anti-C1q antibodies in HUV is controversial: it was associated with renal involvement in the French cohort by Jachiet et al., but showed no prognostic value for the global outcome (ESKD or death) in the present cohort and in the literature review of patients with kidney involvement.
The treatment of HUV and of its kidney involvement is far from established. While corticosteroids were used in all patients from the present cohort, Rituximab was used mostly as a 2nd or 3rd line therapy, but could be proposed as a corticoid-sparing agent in relapsing forms of HUV.
We underline the value of repeat kidney biopsy in these patients, which can allow the differentiation of active versus chronic renal lesions and guide the need for immunosuppressive regimen.
Conclusion
Patients with HUV can display heterogeneous kidney lesions with glomerular, vascular and tubulo-interstitial involvement, with or without immune complex deposits. Kidney involvement of HUV can be severe and relapsing, and lead to ESKD. Corticosteroids, and Rituximab in severe forms, could be the preferential therapeutic options. Screening for proteinuria, and monitoring of blood pressure and serum creatinine could be proposed in all patients with HUV followed-up in Rheumatology or Dermatology, at the time of the diagnosis and during subsequent flares, to diagnose and treat kidney lesions early to avoid chronic kidney damage.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international Chapel Hill consensus conference nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
McDuffie FC, Sams WM, Maldonado JE, Andreini PH, Conn DL, Samayoa EA. Hypocomplementemia with cutaneous vasculitis and arthritis. Possible immune complex syndrome. Mayo Clin Proc. 1973;48(5):340–8.
Schwartz HR, McDuffie FC, Black LF, Schroeter AL, Conn DL. Hypocomplementemic urticarial vasculitis: association with chronic obstructive pulmonary disease. Mayo Clin Proc. 1982;57(4):231–8.
Wisnieski JJ, Baer AN, Christensen J, Cupps TR, Flagg DN, Jones JV, et al. Hypocomplementemic urticarial vasculitis syndrome. Clinical and serologic findings in 18 patients. Medicine (Baltimore). 1995;74(1):24–41.
Jachiet M, Flageul B, Deroux A, Le Quellec A, Maurier F, Cordoliani F, et al. The clinical spectrum and therapeutic management of hypocomplementemic urticarial vasculitis: data from a French nationwide study of fifty-seven patients. Arthritis Rheumatol Hoboken NJ. 2015;67(2):527–34.
Sjöwall C, Mandl T, Skattum L, Olsson M, Mohammad AJ. Epidemiology of hypocomplementaemic urticarial vasculitis (anti-C1q vasculitis). Rheumatology. 2018;57(8):1400–7.
Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P. Acute renal failure – definition, outcome measures, animal models, fluid therapy and information technology needs: the second international consensus conference of the acute Dialysis quality initiative (ADQI) group. Crit Care. 2004;8(4):R204–12.
Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of diet in renal disease study group. Ann Intern Med. 1999;130(6):461–70.
Meyrier A, Français P, Lesavre P, Mougenot B, Ronco P, Jeanmougin M, et al. Hypocomplementemic urticarial vasculitis with glomerulopathy and renal venulitis. Nephrologie. 1984;5(1):1–7.
Ramirez G, Saba SR, Espinoza L. Hypocomplementemic vasculitis and renal involvement. Nephron. 1987;45(2):147–50.
Kobayashi S, Nagase M, Hidaka S, Arai T, Ikegaya N, Hishida A, et al. Membranous nephropathy associated with hypocomplementemic urticarial vasculitis: report of two cases and a review of the literature. Nephron. 1994;66(1):1–7.
Mituiki K, Hirakata H, Oochi N, Nagashima A, Onoyama K, Abe M, et al. Nephrotic syndrome due to membranous glomerulopathy in hypocomplementemic urticarial vasculitis syndrome;--a case report. Nihon Jinzo Gakkai Shi. 1994;36(7):863–70.
Trendelenburg M, Courvoisier S, Späth PJ, Moll S, Mihatsch M, Itin P, et al. Hypocomplementemic urticarial vasculitis or systemic lupus erythematosus? Am J Kidney Dis Off J Natl Kidney Found. 1999;34(4):745–51.
Cadnapaphornchai MA, Saulsbury FT, Norwood VF. Hypocomplementemic urticarial vasculitis: report of a pediatric case. Pediatr Nephrol Berl Ger. 2000;14(4):328–31.
Sessler R, Hasche G, Olbricht CJ. Hypocomplementemic urticarial vasculitis syndrome. Dtsch Med Wochenschr 1946. 2000;125(34–35):1003–6.
Boulay V, Lauque D, Reynaud F, Carles P, Pourrat J. Hypocomplementemic urticarial vasculitis. Presse Medicale Paris Fr 1983. 2000;29(27):1507–9.
Messiaen T, Van Damme B, Kuypers D, Maes B, Vanrenterghem Y. Crescentic glomerulonephritis complicating the course of a hypocomplementemic urticarial vasculitis. Clin Nephrol. 2000;54(5):409–12.
Grimbert P, Schulte K, Buisson C, Desvaux D, Baron C, Pastural M, et al. Renal transplantation in a patient with hypocomplementemic urticarial vasculitis syndrome. Am J Kidney Dis Off J Natl Kidney Found. 2001;37(1):144–8.
el Maghraoui A, Abouzahir A, Mahassine F, Tabache F, Bezza A, Ghafir D, et al. McDuffie hypocomplementemic urticarial vasculitis. Two cases and review of the literature. Rev Med Interne. 2001;22(1):70–4.
Saeki T, Ueno M, Shimada H, Nishi S, Imai N, Miyamura S, et al. Membranoproliferative glomerulonephritis associated with hypocomplementemic urticarial vasculitis after complete remission of membranous nephropathy. Nephron. 2001;88(2):174–7.
Enríquez R, Sirvent AE, Amorós F, Pérez M, Matarredona J, Reyes A. Crescentic membranoproliferative glomerulonephritis and hypocomplementemic urticarial vasculitis. J Nephrol. 2005;18(3):318–22.
Balsam L, Karim M, Miller F, Rubinstein S. Crescentic glomerulonephritis associated with hypocomplementemic urticarial vasculitis syndrome. Am J Kidney Dis Off J Natl Kidney Found. 2008;52(6):1168–73.
Abdallah M, Darghouth S, Hamzaoui S, Ben Ahmed M, Harmel A, Ennafaa M, et al. McDuffie hypocomplementemic urticarial vasculitis associated with Sjögren’s syndrome. Rev Med Interne. 2010;31(7):e8–10.
Ozçakar ZB, Yalçınkaya F, Altugan FS, Kavaz A, Ensari A, Ekim M. Hypocomplementemic urticarial vasculitis syndrome in three siblings. Rheumatol Int. 2013;33(3):763–6.
Al Mosawi ZSA, Al Hermi BEA. Hypocomplementemic urticarial Vasculitis syndrome in an 8-year-old boy: a case report and review of literature. Oman Med J. 2013;28(4):275–7.
Pasini A, Bracaglia C, Aceti A, Vivarelli M, Lavacchini A, Miniaci A, et al. Renal involvement in hypocomplementaemic urticarial vasculitis syndrome: a report of three paediatric cases. Rheumatol Oxf Engl. 2014;53(8):1409–13.
Park C, Choi SW, Kim M, Park J, Lee JS, Chung HC. Membranoproliferative glomerulonephritis presenting as arthropathy and cardiac valvulopathy in hypocomplementemic urticarial vasculitis: a case report. J Med Case Rep. 2014;8:352.
Tanaka M, Moniwa N, Mita T, Tobisawa T, Matsumoto T, Mochizuki A, et al. A case of crescentic glomerulonephritis complicated with Hypocomplementemic urticarial Vasculitis syndrome and ANCA-associated Vasculitis. Case Rep Nephrol Dial. 2017;7(3):144–53.
Gheerbrant H, Giovannini D, Falque L, Andry F, Lugosi M, Deroux A. Severe membranoproliferative glomerulonephritis with polyadenopathy associated with hypocomplementemic urticarial vasculitis syndrome. Presse Medicale Paris Fr 1983. 2017;46(5):547–50.
Jung SW, Choi YY, Choi IS, Kim S, Jeong KH, Song R, et al. Hypocomplementemic urticarial Vasculitis syndrome with membranous nephropathy: case report. J Korean Med Sci. 2017;32(12):2064–8.
Salim SA, Yousuf T, Patel A, Fülöp T, Agarwal M. Hypocomplementemic urticarial Vasculitis syndrome with crescentic glomerulonephritis. Am J Med Sci. 2018;355(2):195–200.
Potlukova E, Kralikova P. Complement component C1q and anti-C1q antibodies in theory and in clinical practice. Scand J Immunol. 2008;67(5):423–30.
Jourde-Chiche N, Daniel L, Chiche L, Burtey S, Mancini J, Jego S, et al. Association between anti-C1q antibodies and glomerular tuft necrosis in lupus nephritis. Clin Nephrol. 2012;77(3):211–8.
Acknowledgements
We thank all physicians participating in the French Study Group on Vasculitis (FSGV) network. We thank Alain Vazi and Laurent Samson for their help with the serum biobank.
Funding
No funding was received for this work.
Author information
Authors and Affiliations
Contributions
AC acquired the data and wrote the manuscript with NJC. MJ, AS, CB, AB, MSD, DiB, AB, OM contributed to collect data on patients’ cases. DaB, NB contributed on laboratory data extraction. BT, XP contributed on patient recruitment through the FVSG network. LD was responsible for renal pathological interpretation. All authors read the final manuscript and approved it.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the principles of the Declaration of Helsinki. An authorization was obtained from the CNIL (Commission Nationale Informatique et Liberté) for the collection of data from patients with renal involvement of HUV (2019_104). For the study on the prevalence of HUV among patients with positive anti-C1q antibodies, sera were obtained from the biobank DC-2012-1704 (Laboratory of Immunology and Department of Nephrology, Hôpital de la Conception, AP-HM, Marseille). All patients had given written informed consent for the use of samples and related clinical data prior to sample collection.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Comparison of patients’ characteristics between those who had positive ANCA at some point during the follow-up, and patients who never had positive ANCA. Supplementary Table 2. Comparison of patients’ characteristics between those who had positive anti-nuclear antibodies at some point during the follow-up, and patients who never had positive anti-nuclear antibodies. Supplementary Table 3. Comparison of patients’ characteristics between those with a favorable outcome (eGFR ≥30 mL/min/1.73m2) at last follow-up and those with an unfavorable outcome (eGFR < 30 mL/min/1.73m2). Supplementary Table 4. Comparison of patients’ characteristics between those with a favorable outcome (eGFR ≥60 mL/min/1.73m2) at last follow-up and those with an unfavorable outcome (eGFR < 60 mL/min/1.73m2). Supplementary Table 5. Comparison of patients’ characteristics between those who relapsed and those who did not relapse during the follow-up.
Additional file 2: Figure S1
. Treatment and renal outcome of the 9 other patients from the present cohort.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Corthier, A., Jachiet, M., Bertin, D. et al. Biopsy-proven kidney involvement in hypocomplementemic urticarial vasculitis. BMC Nephrol 23, 67 (2022). https://doi.org/10.1186/s12882-022-02689-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-022-02689-8