
Abstract
Background
Alterations in the SCN5A gene encoding the cardiac sodium channel Nav1.5 have been linked to a number of arrhythmia syndromes and diseases including long-QT syndrome (LQTS), Brugada syndrome (BrS) and dilative cardiomyopathy (DCM), which may predispose to fatal arrhythmias and sudden death. We identified the heterozygous variant c.316A > G, p.(Ser106Gly) in a 35-year-old patient with survived cardiac arrest. In the present study, we aimed to investigate the functional impact of the variant to clarify the medical relevance.
Methods
Mutant as well as wild type GFP tagged Nav1.5 channels were expressed in HEK293 cells. We performed functional characterization experiments using patch-clamp technique.
Results
Electrophysiological measurements indicated, that the detected missense variant alters Nav1.5 channel functionality leading to a gain-of-function effect. Cells expressing S106G channels show an increase in Nav1.5 current over the entire voltage window.
Conclusion
The results support the assumption that the detected sequence aberration alters Nav1.5 channel function and may predispose to cardiac arrhythmias and sudden cardiac death.
Similar content being viewed by others


Background
Cardiac sodium channels are responsible for the inward depolarizing current (INa) in the initial phase of the cardiac action potential. The cardiac expressed gene SCN5A encodes the pore-forming ion conducting α-subunit of the voltage-gated sodium channel Nav1.5, which is highly conserved among species [1,2,3,4]. The Na+ current resulting from Nav1.5 is fundamental in initiation and propagation of action potentials and is therefore responsible for cardiac excitability and conduction velocity [2]. In cardiomyocytes, Nav1.5 is preferentially localized at intercalated discs and at the T-tubular system [1].
The sodium channel Nav1.5 consists of four homologous domains (DI, DII, DIII and DIV), each consisting of six membrane spanning segments (S1 - S6). The positively charged S4 segments act as voltage sensor and are responsible for the voltage-dependent ion channel activation. The pore of the sodium channel is formed by the extracellular loops (P-loops) between S5 and S6 segments. The N- as well as the C- terminal region of the α-subunit are located in the cytoplasm [3,4,5,6,7].
Genetic aberrations in SCN5A have been associated with different types of arrhythmias and diseases including Brugada syndrome (BrS), long-QT syndrome (LQTS), sick-sinus-syndrome (SSS), atrial fibrillation (AF), dilated cardiomyopathy (DCM) and non-cardiac diseases as for example epilepsy. However, abnormalities in the sodium channel can also lead to overlapping phenotypes, so called overlap syndromes [3, 6, 8,9,10]. Loss-of-function mutations have been associated with Brugada syndrome, whereas gain-of-function variants are known to cause long QT syndrome type 3. Next to those electrical diseases, loss- as well as gain-of-function variants were described in cases of the structural heart disease dilated cardiomyopathy. However, the precise mechanism leading to the DCM phenotype remains unclear [3, 10, 11]. The pathogenesis and clinical spectrum of SCN5A variants is quite complex. Whereas one patient is showing a severe phenotype, another patient carrying the same genetic variant exhibits no clinical conspicuities. Phenotypes may even vary within one family. In the worst case, sudden cardiac death (SCD) is the first sign of a hereditary disease. The variable disease severity, which is most likely influenced by environmental and genetic factors, remains incompletely understood yet [3, 10, 12].
In the present study, we identified the variant of unknown significance c.316A > G, p.(Ser106Gly) in the N-terminal region of the cardiac sodium channel Nav1.5 in a patient with survived cardiac arrest. Variants in the N-terminus of Nav1.5 have been identified in patients with BrS and LQTS. However, the role of the N-terminal region on trafficking, localization and regulation is not fully understood until now [13, 14]. In order to better assess the relevance of the detected variant, we performed functional studies in HEK293 cells.
Methods
Ethics statement
The study was conducted in accordance with the “Declaration of Helsinki”. The patient signed informed consent with approval for publication.
Genetic analysis
DNA was extracted from blood using salting out procedure. Genetic screening was carried out by means of Next-generation sequencing (NGS). Library preparation was performed using Nextera Flex Technology combined with the TruSight Cardio Panel (Illumina) consisting of 174 genes with known cardiac associations (supplementary information). Subsequent sequencing was carried out on a MiniSeq System (Illumina). The resulting reads were aligned to the human reference genome GRCh37/hg19. Variant calling and further evaluation was performed using the Software GensearchNGS (Phenosystems). Variants were filtered according to a pre-established prioritization protocol as described previously [15]. Evaluation was mainly based on presumed functional impact on the protein and allele frequency. Genetic aberrations expected to disrupt protein function and with minor allele frequency below 0.2% in public available databases (The Genome Aggregation and Exome Sequencing Project databases) were further evaluated. Detected sequence variants were assessed using common databases (as e.g. public population databases, Human Gene Mutation Database) and applying in silico prediction tools (PolyPhen-2 [16], MutationTaster [17], SIFT [18], CADD [19]). The identified variant was confirmed by polymerase chain reaction (PCR) and standard direct sequencing by means of BigDye Terminator v1.1 chemistry and 3130xl Genetic Analyzer (Applied Biosystems). The resulting data were evaluated using SeqScape Software.
Mutagenesis
For site-directed mutagenesis, the pcDNA3.1-EGFP vector comprising the wild cDNA sequence of SCN5A (hH1a isoform) was used (kindly provided by N. Neyroud, PhD, Inserm-Sorbonne Université Paris, France). The required sequence variant was induced using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). The resulting constructs were verified by direct sequencing.
Electrophysiological measurements
For the functional characterization of the variant p.S106G, the mutant as well as wild type Nav1.5 channels were expressed in human embryonic kidney (HEK) cells, cell line 293 as described previously [20]. To mimic the heterozygous state of the patient, wildtype and mutant channels were also co-transfected in a 1/1 ratio (WT/MT). HEK293 cells were cultivated at 37 °C, 5% CO2 and passaged by transferring them to 35 mm plates. Transfection was performed by means of TurboFect transfection reagent (Thermo Fisher Scientific, Waltham, USA) and 3 μg of Nav1.5 constructs in the pcDNA3.1-EGFP vector. On the next day, cells were detached using accutase (PAA, GE Health Freiburg, Germany). Four hundred microliters were transferred to a new plate comprising 2 ml medium. Electrophysiological measurements were performed on single cells approximately 36 h following transfection using conventional patch clamp technique. Current measurements were carried out at room temperature in whole-cell configuration. Measurements and data acquisition were performed with an EPC-9 Patch Clamp amplifier (HEKA, Lambrecht, Germany) and the Patch Master (HEKA) software. Data were low pass filtered at 10 kHz and sampled at 20 kHz. Currents were measured in a bath solution composed of 135 mM NaCl, 4 mM KCl, 1 mM CaCl2, 2 mM MgCl2, 20 mM Glucose and 10 mM HEPES (pH 7.4, adjusted with NaOH). The pipette solution consisted of 140 mM CsCl, 5 mM NaCl, 4 mM Mg-ATP, 2 mM MgCl2, 5 mM EGTA, 10 mM HEPES (pH 7.4, adjusted with CsOH). In order to measure the peak current amplitudes and determine current/voltage (I/V) relationships, the cells were clamped from a holding potential of − 140 mV (100 ms) to test voltages ranging from − 100 mV to + 50 mV in 5 mV steps. To analyze the steady-state inactivation, cells were clamped from a holding potential of − 100 mV (100 ms) to test voltages ranging from − 140 mV to − 20 mV by 10 mV increments (500 ms), followed by a 50 ms test pulse to + 20 mV. For data analysis, we utilized Igor Pro 6.03 software (WaveMetrics, Lake Oswego, OR).
For noise analysis, Nav1.5 WT and S106G channels were activated 200 times by a depolarizing voltage step from a holding potential of − 140 mV to − 20 mV or − 40 mV. Current traces were imported into MATLAB to calculate the mean current <I > (t), the current differences between successive traces ΔIn(t), the mean current differences <ΔI>(t), and the variance of the current differences σ2(t). <I > (t), ΔIn(t), <ΔI>(t), and σ2(t) were calculated with the following equations [21]:
where n is trace number and M the total number of traces collected. σ2 values were plotted against Imean and fitted with the following parabolic equation:
where i is the single channel amplitude, N the total number of channels, and σb2 the background noise. Data were fitted with the constrain that σb2 is ≥0.
Data analysis
Results are reported as mean ± standard deviation (SD) of n experiments. An unpaired student’s t-test was applied to determine the statistical significance of the results obtained.
Results
Clinical background and genetic analysis
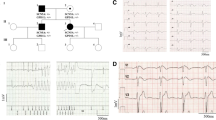
Next-generation sequencing using a defined cardio panel revealed the heterozygous SCN5A variant c.316A > G, p.(Ser106Gly), rs1331765859, in a patient with survived cardiac arrest. This variant is localized in the cytoplasmic N-terminal region of the Nav1.5 channel (Fig. 1a). No further potentially informative rare variant (MAF ≤ 0.2%) was detected. The male patient experienced sudden cardiac arrest at the age of 35 and was finally resuscitated after 53 defibrillations. Cardiological examination revealed no distinct clinical findings and echocardiographic findings most likely pointed towards an incipient dilative cardiomyopathy as the left ventricular ejection fraction was initially slightly reduced (LV-EF 50–55%), but normalized quickly. Hence, this assumption could not be confirmed during follow-up investigations including cardiac magnetic resonance imaging (MRI). ECG tracings did not show specific findings and a provocative test with intravenous administration of the sodium channel blocker ajmaline in order to unmask a concealed Brugada syndrome was performed without displaying a diagnostic Brugada type I-ECG pattern (Fig. 2). So the cause for the cardiac arrest of the patient remained unclear and a arrhythmogenic cardiomyopathy could not be excluded. The patient stated that his father received the diagnosis of a cardiomyopathy, but unfortunately, he was not willing to provide more detailed clinical data and refused genetic analyses. The patient is also father of a toddler, but due to agitation, cardiological examination (ECG) was not possible until now. In order to prevent anxiety no genetic analyses will be performed prior to cardiological assesment due to missing specific cardiological consequences.

Detection of the SCN5A variant p.(Ser106Gly) in a patient with aborted cardiac arrest. a Schematic representation of the Nav1.5 channel and localization of the detected N-terminal variant. b Alignment of a partial N-terminal sequence among mammals. The affected amino acid is indicated in red. c Assessment of the genetic variant by in silico prediction tools

ECG findings of the patient. a ECG taken 1 year previous to the cardiac event is showing sinus rhythm with a heart rate of 66/min, flat P waves, PR interval 140 ms, QRS 100 ms, T inversion in lead III and QTc of 389 ms. b ECG from approximately 3 months after cardiac event under beta-blocker therapy is showing sinus bradycardia with a heart rate of 38/min, flat P waves, PR interval 190 ms, QRS 110 ms and QTc of 365 ms
The amino acid serine (p.Ser106) in the intracellular N-terminal region of the Nav1.5 channel is highly conserved in mammals (Fig. 1b). The sequence variant is not listed in the common databases Exome Variant Server, ExAC (Exome Aggregation Consortium) or GnomAD (The Genome Aggregation Database) indicating that this is a very rare variant. It is only listed in the TOPmed database (Trans-Omics for Precision Medicine). The frequency stated in this database is 0.001% (in 125,568 alleles). Quite recently, the variant has been described in a sudden unexpected death of a 31 year old woman [22]. Aberrations in adjacent regions (p.Arg104Gln; p.Arg104Trp; Asn109Lys) have been associated with Brugada Syndrome [13, 14, 23].
In order to further assess the possible impact of the variant, several in silico predictions were applied. All tools were pointing towards a possible causative impact on the protein (Fig. 1c). Due to missing functional as well as co-segregation studies, this variant was classified as variant of unknown significance (VUS) according to the American College of Medical Genetics (ACMG) standards [24].
Electrophysiological measurements in HEK293 cells
In order to examine the functional relevance of the NaV1.5 variant p.Ser106Gly, wild type (WT) or mutant (MT) Nav1.5 channels were expressed with an n-terminal GFP-tag in HEK293 cells. The representative current traces in Fig. 3a show that whole-cell patch clamp recordings of GFP positive cells exhibit in all cases large, NaV-type, inward currents. When expressed at equivalent levels, the Nav1.5-S106G generated current was higher relative to that of the WT channel (Fig. 3b). At a reference voltage of − 25 mV the current density of the mutant exceeded the control value by a factor of 1.6 (P < 0.05). The density of the Na+ current exceeded that of the control by a factor of 1.3 and fell in between control and mutant when both were transfected together (Fig. 3c). Because of a high variability of the measured current densities this difference is not significant.

Nav1.5 S106G results in gain of function. a Typical Na+ currents in HEK293 cells transiently expressing NaV1.5 WT (top) or NaV1.5-S106G mutant (down). Currents were elicited by clamping cells from holding voltage (− 140 mV, 100 mS) to test voltages (between + 50 mV and − 100 mV, 100 ms) and back to post clamp voltage (− 140 mV, 100 ms). b Mean current density/voltage relationships (±SE) from cells transfected with WT (black, n = 9) or S106G mutant (red, n = 8). c Peak currents at − 25 mV from (b) for WT (black) mutant (red) and data from cells co-transfected with WT and S106G mutant (blue, n = 12). d Boltzmann fits of voltage-dependent inactivation (left) and activation (right) for Nav1.5 WT (black/grey) and NavS106G mutant (red/brown). The mutation causes a positive shift in the V1/2 value for inactivation (− 88.2 ± 0.8 mV versus 91.5 ± 1 mV) and a negative shift of the V1/2 value for activation (− 45.3 ± 0.9 mV versus − 39.9 ± 0.8 mV). The P value for a difference between V1/2 values of the WT and mutant < 0.05. Inset: estimated window currents for WT and mutant from (b). The colors of the line correspond to those in (c)
The S106G mutation also exhibited small effects on the voltage dependency of the channel. The V0.5 value for voltage-dependent inactivation is shifted by 3.3 mV positive relative to the WT channel (− 45.3 mV (N = 8) versus − 39.9 mV (N = 9) respectively). The respective value for voltage dependence activation is in the mutant shifted negative by 5.4 mV (− 91.5 vs. -88.2; P < 0.05 Fig. 3d). Combined with the shift in the inactivation curve this predicts a slight increase and right shift of the window current, which is available during excitation (Fig. 3d). This combination of elevated current density and shift of the voltage dependency of the channel is expected to elevate the Na+ load of cardiomyocytes. We also determined the same parameters for activation and inactivation in conditions in which WT and MT channel were expressed together (Tab. 1). The results show that the curves for activation and inactivation tend towards those of the mutant. They are significantly different (P < 0.05) from the reference values of the WT channel.
The increase in current density of the mutant relative to the WT over the entire voltage spectrum could originate from an increase in unitary conductance or an increase in the number of channels. To examine whether the mutation affects the unitary conductance we estimated the single channel amplitude by non-stationary noise analysis [25] in cells expressing the WT channel, the S106G mutant or both. For the noise analysis we considered the dynamics of channel inactivation from the peak current to the baseline (Fig. 4a). To eliminate unwanted changes in the leak conductance we considered for each isochrone the variance in currents between couples of subsequent voltage steps rather than the variance of the absolute values (see materials and methods). For each cell we plotted the variance values as a function of the mean current. The bell-shaped distribution could be fitted with the parabolic Eq. 5 (an example is reported in Fig. 4b). The first derivative of the function at the current minimum is a good estimate of the single channel amplitude i [25].

S106G mutation does not affect unitary channel conductance. a Exemplary recordings of a HEK293 cell expressing the Nav1.5 WT channel for noise analysis. Channels were activated 200 times by stepping membrane from holding voltage (1–40 mV, 100 ms) to test voltage of − 20 mV (100 ms). Red arrows delimit the part of the traces used for the analysis. b Example of noise analysis from one cell expressing Nav1.5 WT channel. The variance between pulses is plotted as a function of the mean current value. The data were fitted with parabolic function (Eq. 5) yielding the unitary channel conductance from the initial slope of the fit. c Average i values (mean ± SE) for WT and S106G channels at two different voltages: − 20 mV (WT n = 3, black; S106G n = 4, red) and − 40 mV (WT n = 4, black; S106G n = 4, red)
For the WT and S106G construct we performed a noise analysis for clamp steps from holding voltage of − 140 mV to − 20 mV and − 40 mV. The data in Fig. 4 show that the estimated single channel amplitudes (i) and the respective unitary conductance values are all very similar. The results of these experiments imply that the mutation has no impact on the unitary conductance of Nav1.5. This result is not surprising considering that the mutation is far from the pore of the channel.
Discussion
Aberrations in the SCN5A gene have been described to cause disease by altered Nav1.5 structure, function or expression level. Various types of arrhythmias and disorders have been associated in the last decades with alterations in this gene. In our study, we describe a rare SCN5A variant, which might represent a potential risk factor for fatal arrhythmias and sudden cardiac death. In the present case, we detected the N-terminal missense variant p.(Ser106Gly) in a patient with aborted cardiac arrest. During follow-up investigations, the patient did not exhibit distinct clinical conspicuities. Quite recently, this variant has been described in a case of sudden unexpected death in a 31 year old woman [22]. It was known that she suffered from syncope, but showed no ECG abnormalities or diagnostic echocardiographic findings prior to death.
The results of the present experiments support the view that the mutation has an impact on channel function and that this could be causally related to life threatening arrhythmias. The electrophysiological characterization shows that the amino acid replacement in the N-terminus of the channel has a gain of functional impact on the channel. The most obvious functional gain is an increase in steady state current. This is functionally also relevant in a heterozygotic patient because mixed expression of WT and MT still elevates the steady state current relative to the WT channel. The mutation has also small impacts on the voltage dependent parameters with the effect that the window between activation and inactivation is slightly increased and right shifted. In analogy to the effect of other mutants of Nav1.5 [5] the combination of these parameters implies that the Ser106Gly mutation could increase in this manner the influx of Na+ and elevate the Na+ lode of cardiomyocytes. The electrical data do not yet explain the disease phenotypes of the patient carrying the mutant. But they underscore that the mutant causes some aberrant functions, which could be causally linked to heart arrhythmias.
Previous studies have already reported a vast number of mutations in the Nav1.5 channel [5, 26, 27]. The majority of these mutations are localized in the transmembrane domains of the protein, where they affect mostly gating features of the channel. In a second group the mutations are found, like in the present case, in cytosolic domains and associated with signaling or interactions with scaffolding proteins [28]. In the context of recent publications the phenotype of the present mutant may however be better explained by a third category namely mutations in which an interaction of cytosolic domains is important for channel trafficking [13, 14]. A contribution of phosphorylation/dephosphorylation of the critical Ser106 to this mechanism seems not relevant. A bioinformatics analysis suggests that several surrounding residues namely Thr101, Thr108 and Ser115 are potential targets of kinases while Ser106 is not [29].
It is interesting to note that the critical S106G mutation is located between two cationic amino acids R104 and R121 in the Nav1.5 channel [13]. Mutations of these two amino acids exhibit the exact opposite effect of the S106G mutation: they decrease the current density and shift the activation curve positive. As an explanation for the decrease in channel density in the R104W and R121W mutants a quality control mechanism was proposed [13]. According to this mechanism the N-termini of Nav1.5-subunits interact in the ER independent on the critical amino acids. When a mutation in one of the binding partners is mis-folded as a result of a mutation, the protein complex is retained in the secretory pathway and degraded. This quality control in the secretory pathway could reduce trafficking of channel proteins to the plasma membrane. It is tempting to speculate that the S106G mutation has the opposite effect in that it accelerates the quality control mechanism and augments in this manner trafficking of the channels to the plasma membrane. Such an increase in the number of channels would be in agreement with the experimental data, which show an increase in Nav1.5 current over the entire voltage window.
The underlying mechanisms of SCN5A mutations leading to gain-of-function effects are mainly due to abnormalities in channel kinetics. Various studies describe, that gain-of-function variants can cause many individual phenotypes, ranging from LQTS, DCM as well as unclear clinical phenotypes and in the worst case sudden death. Frequently, multiple phenotypes, so called overlap syndromes, are described for one and the same SCN5A mutation, which could also be explained by different electrophysiological properties of the channel in different areas of the heart. Furthermore, it is generally assumed, that there are other unknown mechanisms that are for instance related to age, co-morbidities or environmental influences that contribute to genotype-phenotype interactions. Identification of other modulators and genetic factors may help to understand how alterations in SCN5A can lead to so many diverse clinical phenotypes [3, 12, 26].
Based on the electrophysiological data, the severe cardiac event of the patient, in whom the variant was detected and last, the fact that the variant was identified in a further sudden death case of a young woman, who did not show any ECG abnormalities prior to death, conspicuously [22], the n-terminal variant has to be considered as a risk factor for fatal arrhythmias and sudden death.
Conclusions
In the present study, we describe an electrophysiological characterization of the very rare sequence variant Ser106Gly in the SCN5A gene leading to a gain-of-function impact on the sodium channel Nav1.5. As described in previous studies, our data underline the assumption that the cytosolic n-terminal domain is probably important for channel trafficking. The results indicate, that the exchange of Ser for Gly is not recognized by any quality control mechanism and that the mutant is transported to the membrane in a preferred manner.
Availability of data and materials
The raw data are available upon request from the corresponding author. The genetic variant detected during this study is available in the NCBI dbSNP database, rs1331765859. The human reference genome GRCh37/hg19 was used as reference data set in this study.
Abbreviations
- AF:
-
Atrial fibrillation
- BrS:
-
Brugada syndrome
- DCM:
-
Dilated Cardiomyopathy
- ECG:
-
Electrocardiogram
- EGFP:
-
Enhanced green fluorescent protein
- HEK293:
-
Human embryonic kidney cells (cell line 293)
- LQTS:
-
Long QT syndrome
- LV-EF:
-
Left ventricular ejection fraction
- MRI:
-
Magnetic resonance imaging
- MT:
-
Mutant
- NGS:
-
Next-generation sequencing
- SCD:
-
Sudden cardiac death
- SSS:
-
Sick sinus syndrome
- WT:
-
Wild type
References
Rook MB, Evers MM, Vos MA, et al. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc Res. 2012;93:12–23. https://doi.org/10.1093/cvr/cvr252.
Amin AS, Asghari-Roodsari A, Tan HL. Cardiac sodium channelopathies. Pflugers Arch. 2010;460:223–37. https://doi.org/10.1007/s00424-009-0761-0.
Wilde AAM, Amin AS. Clinical spectrum of SCN5A mutations: long QT syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin Electrophysiol. 2018;4:569–79. https://doi.org/10.1016/j.jacep.2018.03.006.
Cohen SA, Barchi RL. Cardiac sodium channel structure and function. Trends Cardiovasc Med. 1992;2:133–40. https://doi.org/10.1016/1050-1738(92)90020-S.
Kass RS. Sodium channel inactivation in heart: a novel role of the carboxy-terminal domain. J Cardiovasc Electrophysiol. 2006;17:S21–5. https://doi.org/10.1111/j.1540-8167.2006.00381.x.
Abriel H. Roles and regulation of the cardiac sodium channel Na v 1.5: recent insights from experimental studies. Cardiovasc Res. 2007;76:381–9. https://doi.org/10.1016/j.cardiores.2007.07.019.
Remme CA. Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J Physiol. 2013;591:4099–116. https://doi.org/10.1113/jphysiol.2013.256461.
McNair WP, Ku L, Taylor MRG, et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;11015:2163–7. https://doi.org/10.1161/01.CIR.0000144458.58660.BB.
Benson DW, Wang DW, Dyment M, et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest. 2003;112:1019–28. https://doi.org/10.1172/JCI200318062.
Veerman CC, Wilde AAM, Lodder EM. The cardiac sodium channel gene SCN5A and its gene product NaV1.5: role in physiology and pathophysiology. Gene. 2015;573:177–87. https://doi.org/10.1016/j.gene.2015.08.062.
Zaklyazminskaya E, Dzemeshkevich S. The role of mutations in the SCN5A gene in cardiomyopathies. Biochim Biophys Acta. 1863;2016:1799–805. https://doi.org/10.1016/j.bbamcr.2016.02.014.
Verkerk AO, Amin AS, Remme CA. Disease modifiers of inherited SCN5A channelopathy. Front Cardiovasc Med. 2018;5:137. https://doi.org/10.3389/fcvm.2018.00137.
Clatot J, Ziyadeh-Isleem A, Maugenre S, et al. Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Na(v)1.5 α-subunits. Cardiovasc Res. 2012;961:53–63. https://doi.org/10.1093/cvr/cvs211.
Gütter C, Benndorf K, Zimmer T. Characterization of N-terminally mutated cardiac Na+ channels associated with long QT syndrome 3 and Brugada syndrome. Front Physiol. 2013;4:153. https://doi.org/10.3389/fphys.2013.00153.
Köffer J, Scheiper-Welling S, Verhoff MA, et al. Post-mortem genetic investigation of cardiac disease-associated genes in sudden infant death syndrome (SIDS) cases. Int J Legal Med. 2020. https://doi.org/10.1007/s00414-020-02394-x.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. https://doi.org/10.1038/nmeth0410-248.
Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. https://doi.org/10.1038/nprot.2009.86.
Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. https://doi.org/10.1038/ng.2892.
Scheiper S, Hertel B, Beckmann BM, et al. Characterization of a novel KCNJ2 sequence variant detected in Andersen-Tawil syndrome patients. BMC Med Genet. 2017;18:113. https://doi.org/10.1186/s12881-017-0472-x.
Liu C, Xie C, Grant K, Su Z, Gao W, Liu Q, Zhou L. Patch-clamp fluorometry-based channel counting to determine HCN channel conductance. J Gen Physiol. 2016;148:65–76.
Hellenthal N, Gaertner-Rommel A, Klauke B, et al. Molecular autopsy of sudden unexplained deaths reveals genetic predispositions for cardiac diseases among young forensic cases. Europace. 2017;19:1881–90. https://doi.org/10.1093/europace/euw247.
Amin AS, de Groot EAA, Ruijter JM, et al. Exercise-induced ECG changes in Brugada syndrome. Circ Arrhythm Electrophysiol. 2009;2:531–9. https://doi.org/10.1161/CIRCEP.109.862441.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. https://doi.org/10.1038/gim.2015.30.
Alvarez O, Gonzalez C, Latorre R. Counting channels: a tutorial guide on ion channel fluctuation analysis. Adv Physiol Educ. 2002;26:327–41. https://doi.org/10.1152/advan.00006.2002.
Detta N, Frisso G, Salvatore F. The multi-faceted aspects of the complex cardiac Nav1.5 protein in membrane function and pathophysiology. Biochim Biophys Acta. 2015;1854:1502–9. https://doi.org/10.1016/j.bbapap.2015.07.009.
Moreau A, Chahine M. A new cardiac channelopathy: from clinical phenotypes to molecular mechanisms associated with Nav1.5 gating pores. Front Cardiovasc Med. 2018;5:139. https://doi.org/10.3389/fcvm.2018.00139.
Musa H, Kline CF, Sturm AC, et al. SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc Natl Acad Sci U S A. 2015;112:12528–33. https://doi.org/10.1073/pnas.1516430112.
Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–20.
Acknowledgements
We thank Isabelle Feinauer for her technical work during cloning experiments.
Funding
This work was in part supported by grant from DLR (50 WB1838) to GT. SK is supported by the Friedrich Flick Foundation. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
SS-W designed the study, performed and evaluated genetic analyses and mutagenesis, interpreted the results and wrote the manuscript draft. PZ performed functional experiments, interpreted and visualized functional data and was involved in writing the manuscript. OR analyzed and interpreted data. B-MB examined the clinical data of the patient. CG was involved in genetic analyses. AM and GT interpreted data and wrote manuscript draft. SK supervised the study and was involved in writing the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The current study was approved by the ethical commission of the University Hospital, Goethe University Frankfurt (protocol number E84/06). Written informed consent to participate was obtained from the patient involved in this study.
Consent for publication
Consent for publication of the clinical data was obtained from all of the participants.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
List of genes included in the TruSight Cardio Panel (Illumina).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Scheiper-Welling, S., Zuccolini, P., Rauh, O. et al. Characterization of an N-terminal Nav1.5 channel variant – a potential risk factor for arrhythmias and sudden death?. BMC Med Genet 21, 227 (2020). https://doi.org/10.1186/s12881-020-01170-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01170-3