
Abstract
Background
Pulmonary hypertension (PH) remains one of the rarest and deadliest diseases. Pulmonary Capillary Hemangiomatosis (PCH) is one of the sub-classes of PH. It was identified using histological and molecular tools and is characterized by the proliferation of capillaries into the alveolar septae. Mutations in the gene encoding the eukaryotic translation initiation factor 2 alpha kinase 4 (EIF2AK4) have recently been linked to this particular subgroup of PH.
Methods
In our effort to unveil the genetic basis of idiopathic and familial cases of PH in Lebanon, we have used whole exome sequencing to document known and/or novel mutations in genes that could explain the underlying phenotype.
Results
We showed bi-allelic mutations in EIF2AK4 in two non-consanguineous families: a novel non-sense mutation c.1672C > T (p.Q558*) and a previously documented deletion c.560_564drlAAGAA (p.K187Rfs9*). Our histological analysis coupled with the CT-scan results showed that the two patients with the p.Q558* mutation have PH. In contrast, only one of the individuals harboring the p.K187Rfs9* variant has a documented PCH while his older brother remains asymtomatic. Differential analysis of the variants in the genes of the neighboring network of EIF2AK4 between the two siblings identified a couple of interesting missense mutations that could account for this discrepancy.
Conclusion
These findings represent a novel documentation of the involvement of EIF2AK4 in the different aspects of pulmonary hypertension. The absence of a molecular mechanism that relates the abrogated function of the protein to the phenotype is still a major hurdle in our understanding of the disease.
Similar content being viewed by others


Background
Pulmonary capillary hemangiomatosis (PCH) is a rare but increasingly recognized cause of pulmonary hypertension [1]. Although it was first described in 1978, fewer than 50 cases have been reported in the literature since [2]. The median survival is 3 years and the only effective therapy is lung transplantation. It is now classified in the diagnostic group 1′, a distinct subgroup of pulmonary arterial hypertension. PCH is a diffuse process characterized by the proliferation of capillaries into the alveolar septae, often invading small vessels and bronchi [3]. It is combined in classification with pulmonary veno-occlusive disease (PVOD) [4]. Indeed, PCH and PVOD overlap in several histopathological findings such as venous obstruction, arterial intimal fibrosis and interlobular septal thickening. They also share clinical, radiological, and hemodynamic characteristics [5, 6].
Patients with PCH present with nonspecific symptoms of dyspnea, fatigue, cough, and hemoptysis. Echocardiogram and right heart catheterisation show changes compatible with pulmonary arterial hypertension (PAH) and similar to those found in other forms of PAH. High resolution CT scans of the chest show centrilobular ground glass opacities, septal lines, and mediastinal lymph node enlargement, findings common to PVOD [2]. Despite clinical suspicion of PCH, a definitive diagnosis requires histological examination of lung tissue and is often delayed until lung transplant and the examination of explanted tissue. Treatment of PCH with PAH-specific therapy has been described as a bridge to lung transplant [7]. Response to PAH-specific therapy is limited. Drug initiation and titrations can induce pulmonary edema and should be performed with caution [2, 7,8,9].
Sporadic and familial cases of PCH have been reported, indicating a genetic component in the etiology of the disease. Mutations in the EIF2AK4 gene (coding for eukaryotic translation initiation factor 2α Kinase 4) were identified as the genetic predisposition causes of PCH. These mutations were first found in an autosomal recessively inherited PCH familial case and in 20% of sporadic cases [10]. EIF2AK4 mutations are also found in PVOD patients, reinforcing the link of these two diseases to a common genetic risk factor [11,12,13].
We report a new EIF2AK4 mutation type in a histologically proven case of PCH. It is also the first reported case of EIF2AK4 mutation in PCH in the Eastern Mediterranean region (Lebanese population). We also report a novel EIF2AK4 homozygous mutation in a family clinically diagnosed with HPAH.
Methods
Subjects and clinical characteristics
Patients and their family members when available were recruited as part of a clincical and genetic research study on PAH in Lebanon between 2015 and 2017. They were eligible for enrolment if the patients had a pulmonary arterial pressure (mPAP) > 25 mmHg at rest and a pulmonary artery wedge pressure (PAWP) < 15 mmHg.
The study was approved by the Institutional Review Board (IRB) at the American University of Beirut Medical Center (AUBMC) (Protocol Number IM.IB.01). Written consent forms were collected from all participants. The collected data include medical history, physical exam, family history for PAH, echocardiography, pulmonary function test, chest computerized tomography (CT), CT pulmonary angiography, ventilation perfusion scan, and pro-Brain Natriuritic Peptide (BNP) blood levels.
Genetic studies
Five millileters of peripheral blood were collected from the patients. We used the Qiagen QIAamp blood midi-kit to extract DNA and we assessed its purity using Nanodrop (Thermo Fisher) at the molecular core facility at AUB. Library preparation and subsequent exome capture was done on an Illumina HiSeq2500 platform at Macrogen (South Korea) using the V5 SureSelect kits (Agilent). Sequences were aligned to the hg19 human genome using Novoalign and variants were called by the Genome Analysis Toolkit (GATK). We used the variant call software (Illumina) to further annotate and analyze the obtained variants. Sanger sequencing was carried on with specific primers to confirm the mutations in the affected and no-affected individuls using the ABI3500 platform at AUB.
Variant analysis
Analysis was performed based on a filtering panel consisting of genes (Additional file 4: Table S1) implicated in PAH as previously described [14]. Only non-synonymous, insertion/deletions variants in the coding regions, and splicing variants with allele frequencies inferior to 1% were filtered in according to their evolutionary conservation, and location within the encoded protein. These criteria were evlauted by in silico predictive software (SIFT: https://sift.bii.a-star.edu.sg/ and Polyphen2: http://genetics.bwh.harvard.edu/pph2/) and scored accordingly (Additional file 1: Figure S1).
Results
A sporadic case of PCH with a previously documented EIF2AK4 variant
The indexed case is a 47-year-old man, non-smoker, who presented to the hospital for progressive dyspnea on exertion of 1 year duration (Fig. 1a). The dyspnea’s onset was insidious, progressed rapidly and affected his daily activity. He reported mild lower extremity edema, but no orthopnea, no paroxysmal nocturnal dyspnea (PND), no wheezing, and no cough. CT angiography of the chest showed no pulmonary embolism nor thickening of interlobular septae (Fig. 1b). The transthoracic echocardiogram showed normal left ventricular function with a moderately dilated right ventricle and a pulmonary arterial systolic pressure of 75 mmHg. Spirometry was also performed and showed no flow obstruction and normal FVC (Functional Vital Capacity). Cardiac MRI showed no evidence of shunt or infiltrative cardiomyopathy. Right heart catheterization showed severe Pulmonary Artery Hypertension (PAH) with a mean pulmonary artery pressure (mPAP) of 68 mmHg and pulmonary wedge pressure of 13 mmHg. The pulmonary vascular resistance was calculated to be 13 Wu. The 6-min-walk was 368 m. An open lung biopsy was performed and it showed that the alveolar septae were thickened with prominent intimal fibrosis and medial thickening of the veins (Fig. 2). Apart from the presence of rare interstitial lymphocytes and mildly increased alveolar macrophages, there was no significant inflammation. These findings confirmed the suspicion of PCH. The patient was started on Sildenafil and the dose was slowly titrated to 25 mg TID. He experienced only mild improvement, which prompted the addition of Bosentan to alleviate the symptoms. He continued to deteriorate over the next 2 years leading to his death 3 years after the initial diagnosis.

Clinical and genetic characterization of Family A patients. a Pedigree for the family, showing the frameshift stop-gained EIF2AK4 mutation. The −/− symbol is for normal genes, −/+ is for heterozygous mutations and the +/+ is for homozygous mutations. Male; Circle, female; open symbol, unaffected; filled symbol, affected, the symbol with arrow: Death. Current age or age at death in years is between parenthesis. b The chest CT scan of the index patient (AII.2) with a clinical presentation of Pulmonary veno-occlusive disease shows dilated pulmonary trunk, small pleural, pericardial effsuions, and interlobular septal thickening in the lung bases. The lower panel is the asymptomatic older brother’s (AII.1) CT scan which doesn’t show any abnormality in the chest. c Sanger chromatogram tracing confirming the NGS variant sequence for both AII.1 and AII.2, and showing the EIF2AK4 mutation as detailed in the supp. Table from whole exome sequencing results

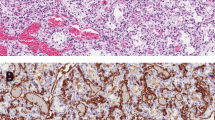
Histopathologic findings on open lung biopsy of patient AII.2. The alveolar septa are highly thickened (a). Small and intermediate size veins show thickened walls and narrowed lumens due to concentric laminar fibrosis (a and b respectively). Medium and large arteries have a thickened media and prominent intimal fibrosis (c,d). Symbols: *, intima; arrow, media; arrow head, alveolar septa
Whole exome sequencing was perfomed on the patient, and after filtering in variants with a minor allele frequency (MAF) < 1%, a bi-allelic mutation in EIF2AK4 was detected. The p.K187 fs mutation (rs772487425) was previously reported in a 32-year-old female with a documented histological PVOD phenotype [12, 15]. In our case, the patient had no family history of pulmonary hypertension and had two siblings. Sanger sequencing confirmed that the patient (AII.2) had the homozygous mutation along with his older brother (AII.1) (Fig. 1c). His sister (AII.3) and mother (AI.2) were heterozygous for the mutation. The father (AI.1) did not consent to participate in the study. Clinical workout of AII.1 showed, at 50 years of age, no echocardiographic findings suggestive of pulmonary arterial hypertension. A CT scan of the chest was normal with no centrilobular ground glass opacities, no septal lines, and no mediastinal lymph node enlargement which would have suggested that he had PCH or PVOD like his brother (Fig. 1b). His pulmonary function test was normal. This prompted us to further cross-compare the results of the exome sequencing between the two siblings with bi-allelic EIF2AK4 mutation in order to account for the discrepancy in the phenotype. We based our hypothesis on a potential variant that would have “a gain of function” effect on the neighbouring network of genes around EIF2AK4 (Additional file 2: Figure S2). None of the other EIF gene encoding kinases displayed a differential variant signature between AII.1 and AII.2 (Additional file 5: Table S2). Interestingly, only one missense variant p.V682 M in the gene encoding GCN1L1 (general control of amino acid synthesis-1 like protein 1), an EIF2AK4 obligate partner was detected in the unaffected individual with a minor allele frequency of 0 (Additional file 6: Table S3).
A familial case of pulmonary hypertension with a novel mutation in EIF2AK4
The indexed patient BII.4 is a woman who presented at the age of 36 years with slowly progressing dyspnea on exertion of 1 year duration (Fig. 3a). Her diagnostic work up started with a CT angiography that showed a dilated pulmonary artery suggestive of PAH, normal parenchyma, and no pulmonary embolism. Transthoracic echocardiogram showed normal left ventricular function with tricuspid regurgitation and an estimated pulmonary artery systolic pressure of 90 mmHg. The mPAP by right heart catheter was 45 mmHg. She was diagnosed with idiopathic PAH and started on Sildenafil with good response. She remained stable for 7 years, at which time Bosentan was added. She remainded stable for another 7 years, at which time she developed severe community acquired pneumonia and ARDS and passed away at the age of 50 years. 6 years following her initial diagnosis, her brother (BII.1) presented with similar symptoms at the age of 51, while the parents did not participate in the study. The brother’s mPAP was 50 mmHg and he was started on a Sildenafil-Bosentan combination therapy and remained stable 8 years later with good functional status. Like his sister, patient BII.1 had good response to PAH targeted therapy with no episodes of congestion upon the initiation of the vasodilators. Once we found the novel homozygous nonsense – p.Q558*- mutation in E1F2AK4 in both patients (Fig. 3c), high resolution chest CTs were repeated and reviewed. Both patients had no radiological changes suggestive of PVOD or PCH (Fig. 3b).

Clinical and genetic characterization of Family A patients. a Pedigree for the family, showing the frameshift stop-gained EIF2AK4 mutation. The −/− symbol is for normal genes, −/+ is for heterozygous mutations and the +/+ is for homozygous mutations. Male; Circle, female; open symbol, unaffected; filled symbol, affected, the symbol with arrow: Death. Current age or age at death in years is between parenthesis. b The chest CT scan of the index patient (BII.4) with a clinical presentation of hereditary pulmonary arterial hypertension shows normal parenchyma and minimal pericardial effusion. The lower panel is the older brother’s (BII.1) CT scan which shows enlarged pulmonary artery and no parenchymal changes. Both Images did not have neither pleural effusion, nor centrilobular nodules. c Sanger chromatogram tracing confirming the NGS variant sequence for the patient and showing the mutation as detailed in the supp. Table from whole exome sequencing results
Discussion
This is the first documentation of EIF2AK4 variants in Lebanon, a small country in the Eastern Mediterranean region with consanguineous marriage rates that remain relatively high. On one hand, we have a patient with a previously reported EIF2AK4 mutation presenting as a sporadic PCH case confirmed by histological findings and radiological profile while his two-years older brother who carries the same mutation has no signs or symptoms of the disease. On the other, we describe for the first time a family presenting with a novel homozygous EIF2AK4 mutation with a confirmed clinical picture of hereditary PAH.
PCH/PVOD is a rare cause of precapillary pulmonary hypertension and can mistakenly be diagnosed as idiopathic PAH. Diagnosis of PCH is based on clinical suspicion. High resolution CT scan findings of centrilobular ground glass opacities, subpleural thickened septal lines, and mediastinal lymphadenopathy, can provide clues for the diagnosis [1, 2]. Prevalence of biallelic EIF2AK4 mutation is 9% in the sporadic group of patients and may help, when present, to confirm PCH/PVOD diagnosis by performing genetic testing and avoiding a more risky confirmation by biopsy [2, 5]. Our first patient presented at age 46 and had advanced New York Heart Association (NYHA) functional class on presentation which is common among patients with PCH/PVOD. The diagnosis of PCH was confirmed by histologic examination of an open lung biopsy. He had mild improvement of his symptoms with PAH-targeted therapy of PDE5 inhibitor and Endothelin Receptor Antagonist (ERA) along with diuretics. He did not develop pulmonary edema after the initiation of therapy which can happen in PCH/PVOD patients [2].
The p.K187Rfs mutation in EIF2AK4 detected in our second patient was previously reported in a patient diagnosed at the age of 32 with PCH/PVOD [12, 15]. Our patient was much older at the onset of diagnosis and his older brother (now 51) remains unaffected. Our patient, and his unaffected brother, had no exposure to solvent or chemotherapy, which are reported causes of PCH/PVOD [9, 16]. These results suggest that the “penetrance” of the mutations in EIF2AK4 is similar to other familial cases of PAH whereby mutations in BMPR2 are partially penetrant [17,18,19], and/or that environmental factors including diet could be implicated in the differential phenotype observed so far [8, 15]. Previous studies have argued that mutations in EIF2AK4 lead either to a defective protein or to the absence of expression of the protein. However, there are no functional studies so far to assess the effect of missense mutations or truncated protein products on the activity of the encoded EIF2AK4 protein despite a recent attempt to quantify the protein’s expression in postmortem lung tissues [20]. It is even perplexing that the Eif2ak4−/− mouse model did not show any substantial phenotype pertaining to PCH/PVOD [21]. The explanation could be related to a genetic/genomic background “elevated potential “that compensates for the abrogated activity of the mutated protein.
It is clear that little is known with respect to the EIF2AK4 mutation’s penetrance due to the rare disease and difficult diagnosis. There are suggestions to the fact that even heterozygous compound mutations might manifest as PH supporting a “second hit” phenomenon [22]. We have selected to overview some of the genes in the close vicinity of the EIF2AK4 network (Additional file 2: Figure S2). There are three additional enzymes that could phosphorylate the elongation initiation factor type 2 (EIF2a) encoded by the genes EIF2AK1–3; none showed differential variants between the two siblings (Additional file 5: Table S2). On the contrary the EIF2AK4 activating cofactor GCN1L1 was shown to harbor a missense variation only in the phenotypically normal individual. This variant is unique since it is not found in the GNOmad exomes and genomes and is predicted to have a mild effect on protein function. We hypothesize that this variant would confer a gain of function activity that enhances its interaction with EIF2AK4, although the domain of interaction with the latter is situated in the C-terminal region, which is deleted in the presumably truncated protein [23, 24]. Alternatively, a different diet regimen may be responsible for the “phenotypic” difference between the siblings, taking into consideration that EIF2AK4 has been shown to prevent oxiditative damage caused by an amino-acid unbalanced diet [25, 26]. Finally, additional variants in genes with no documented implication in PCH/PVOD could also account for this discrepancy. Clinical follow up is needed for the individual at risk as the phenotype expression could appear later, as was the case in the founder EIF2AK4 mutation in the Iberian Romani population [11].
Conclusion
Despite the breakthrough in the identification of EIF2AK4 as a major player in PVOD/PCH, little is known of its implication in PAH, and much of the controversy linked to this issue is related to the proper radiological and clinical evidence that supports or refutes the causal relationship. The first report of a mutation in EIF2AK4 reponsible for hereditary pulmonary arterial hypertension came as a combined heterozygous mutation in conjunction with a BMPR2 mutation [22]. Since then, few large cohort studies have shown hereditary pulmonary arterial hypertension bi-allelic mutations in EIF2AK4. These mutations were also described in 1% of patients presenting with a clinical diagnosis of idiopathic PAH. The patients had a worse prognosis when compared with other PAH patients within the same cohort. They tended to have more radiological findings that are found in PCH/PVOD than PAH patients without the mutation. This was also the case in the BRIDGE study whereby 9 patients out of 864 with either idiopathic PAH or hereditary PAH had bi-allelic EIF2AK4 mutations [5]. Our patients with the novel homozygous EIF2AK4 mutation p.Q558* were clinically diagnosed with hereditary PAH, and had no findings suggestive of PCH/PVOD. The presence of the biallelic mutation was the first clue to a possible PCH diagnosis. Unlike patients with PCH/PVOD, they both had good response to vasoldilators therapy with improvement in their functional status. In contrast to the rapidly progressive nature of PCH/PVOD, the disease seemed to remain stable for an extended period of time. The chest CT of our patients did not show interlobular septal thickening as is the case in previous studies of PAH patients with bi-allelic mutations in EIF2AK4 [5, 10, 27]. These genetic, clinical, and radiological findings raise the question about the functional properties of the mutated EIF2AK4 protein products and whether specific domains of the protein are implicated in divergent signaling pathways. Indeed, our compilation of the described mutations in EIF2AK4 causing PCH/PVOD or PAH show a clustering of mutations in the His-tRNA synthase like domain of the protein for patients with PCH/PVOD (Additional file 3: Figure S3). Structure-function studies are warranted to delineate the phenotype/genotype correlations and better understand the molecular pathways involved in pulmonary hypertension.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Exome sequencing files are available to share through a direct request process to the corresponding authors.
Abbreviations
- ACE:
-
Angiotensin converting enzyme inhibitors
- ARDS:
-
Acute respiratory distress syndrome
- AUB:
-
American University of Beirut
- AUBMC:
-
American University of Beirut Medical Center
- BMPR2:
-
Bone morphogenetic protein receptor type 2
- BNP:
-
Brain Natriuretic Peptide
- BNP:
-
Pro-B-type natriuretic peptide
- CT:
-
Computerized tomography
- EIF:
-
Eukaryotic initiation factor
- EIF2a :
-
Elongation initiation factor type 2
- EIF2AK1–3:
-
Eukaryotic translation initiation factor 2 alpha kinase 1–3
- EIF2AK4:
-
Eukaryotic translation initiation factor 2 alpha kinase 4
- ERA:
-
Endothelin Receptor Antagonist
- GATK:
-
Genome Analysis Toolkit
- GCN1L1:
-
General control of amino acid synthesis-1 like protein 1
- gnomAD:
-
Genome Aggregation Database
- HPAH:
-
Heritable pulmonary arterial hypertension
- IPAH:
-
Idiopathic pulmonary arterial hypertension
- IRB:
-
Institutional review board
- MAF:
-
Minor allele frequency
- mPAP:
-
Mean pulmonary arterial pressure
- MRI:
-
Magnetic resonance imaging
- PAH:
-
Pulmonary arterial hypertension
- PAP:
-
Pulmonary arterial pressure
- PAWP:
-
Pulmonary artery wedge pressure
- PCH:
-
Pulmonary capillary hemangiomatosis
- PDE5:
-
Phosphodiesterase type 5
- PDGFs:
-
Platelet-derived growth factors
- PH:
-
Pulmonary hypertension
- PolyPhen-2:
-
Polymorphism Phenotyping v2
- PVOD:
-
Pulmonary veno-occlusive disease
- SIFT:
-
Sorting Intolerant from Tolerant
- SNP:
-
Single Nucleotide Polymorphism
- TID:
-
Three times a day
- VEGFs:
-
Vascular endothelial growth factors
- WHO:
-
World Health Organization’s
- WU:
-
Wood Unit
References
Montani D, Achouh L, Dorfmuller P, Le Pavec J, Sztrymf B, Tcherakian C, Rabiller A, Haque R, Sitbon O, Jais X, et al. Pulmonary veno-occlusive disease: clinical, functional, radiologic, and hemodynamic characteristics and outcome of 24 cases confirmed by histology. Medicine (Baltimore). 2008;87(4):220–33.
Montani D, Lau EM, Dorfmuller P, Girerd B, Jais X, Savale L, Perros F, Nossent E, Garcia G, Parent F, et al. Pulmonary veno-occlusive disease. Eur Respir J. 2016;47(5):1518–34.
Wagenvoort CA, Beetstra A, Spijker J. Capillary haemangiomatosis of the lungs. Histopathology. 1978;2(6):401–6.
Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Rev Esp Cardiol (Engl Ed). 2016;69(2):177.
Hadinnapola C, Bleda M, Haimel M, Screaton N, Swift A, Dorfmuller P, Preston SD, Southwood M, Hernandez-Sanchez J, Martin J, et al. Phenotypic characterization of EIF2AK4 mutation carriers in a large cohort of patients diagnosed clinically with pulmonary arterial hypertension. Circulation. 2017;136(21):2022–33.
Chaisson NF, Dodson MW, Elliott CG. Pulmonary capillary Hemangiomatosis and pulmonary Veno-occlusive disease. Clin Chest Med. 2016;37(3):523–34.
Montani D, Jais X, Price LC, Achouh L, Degano B, Mercier O, Mussot S, Fadel E, Dartevelle P, Sitbon O, et al. Cautious epoprostenol therapy is a safe bridge to lung transplantation in pulmonary veno-occlusive disease. Eur Respir J. 2009;34(6):1348–56.
Girerd B, Montani D, Jais X, Eyries M, Yaici A, Sztrymf B, Savale L, Parent F, Coulet F, Godinas L, et al. Genetic counselling in a national referral Centre for pulmonary hypertension. Eur Respir J. 2016;47(2):541–52.
Ranchoux B, Gunther S, Quarck R, Chaumais MC, Dorfmuller P, Antigny F, Dumas SJ, Raymond N, Lau E, Savale L, et al. Chemotherapy-induced pulmonary hypertension: role of alkylating agents. Am J Pathol. 2015;185(2):356–71.
Best DH, Sumner KL, Smith BP, Damjanovich-Colmenares K, Nakayama I, Brown LM, Ha Y, Paul E, Morris A, Jama MA, et al. EIF2AK4 mutations in patients diagnosed with pulmonary arterial hypertension. Chest. 2017;151(4):821–8.
Navas Tejedor P, Palomino Doza J, Tenorio Castano JA, Enguita Valls AB, Rodriguez Reguero JJ, Martinez Menaca A, Hernandez Gonzalez I, Bueno Zamora H, Lapunzina Badia PD, Escribano Subias P. Variable expressivity of a founder mutation in the EIF2AK4 gene in hereditary pulmonary Veno-occlusive disease and its impact on survival. Rev Esp Cardiol (Engl Ed). 2018;71(2):86–94.
Eyries M, Montani D, Girerd B, Perret C, Leroy A, Lonjou C, Chelghoum N, Coulet F, Bonnet D, Dorfmuller P, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet. 2014;46(1):65–9.
Tenorio J, Navas P, Barrios E, Fernandez L, Nevado J, Quezada CA, Lopez-Meseguer M, Arias P, Mena R, Lobo JL, et al. A founder EIF2AK4 mutation causes an aggressive form of pulmonary arterial hypertension in Iberian gypsies. Clin Genet. 2015;88(6):579–83.
Abou Hassan OK, Haidar W, Nemer G, Skouri H, Haddad F, BouAkl I. Clinical and genetic characteristics of pulmonary arterial hypertension in Lebanon. BMC Med Genet. 2018;19(1):89.
Machado RD, Southgate L, Eichstaedt CA, Aldred MA, Austin ED, Best DH, Chung WK, Benjamin N, Elliott CG, Eyries M, et al. Pulmonary arterial hypertension: a current perspective on established and emerging molecular genetic defects. Hum Mutat. 2015;36(12):1113–27.
Perros F, Gunther S, Ranchoux B, Godinas L, Antigny F, Chaumais MC, Dorfmuller P, Hautefort A, Raymond N, Savale L, et al. Mitomycin-induced pulmonary Veno-occlusive disease: evidence from human disease and animal models. Circulation. 2015;132(9):834–47.
Garcia-Rivas G, Jerjes-Sanchez C, Rodriguez D, Garcia-Pelaez J, Trevino V. A systematic review of genetic mutations in pulmonary arterial hypertension. BMC Med Genet. 2017;18(1):82.
Best DH, Austin ED, Chung WK, Elliott CG. Genetics of pulmonary hypertension. Curr Opin Cardiol. 2014;29(6):520–7.
Hamid R, Cogan JD, Hedges LK, Austin E, Phillips JA 3rd, Newman JH, Loyd JE. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum Mutat. 2009;30(4):649–54.
Nossent EJ, Antigny F, Montani D, Bogaard HJ, Ghigna MR, Lambert M. Thomas de Montpreville V, Girerd B, Jais X, Savale L et al: Pulmonary vascular remodeling patterns and expression of general control nonderepressible 2 (GCN2) in pulmonary veno-occlusive disease. J Heart Lung Transplant. 2018;37(5):647–55.
Anthony TG, McDaniel BJ, Byerley RL, McGrath BC, Cavener DR, McNurlan MA, Wek RC. Preservation of liver protein synthesis during dietary leucine deprivation occurs at the expense of skeletal muscle mass in mice deleted for eIF2 kinase GCN2. J Biol Chem. 2004;279(35):36553–61.
Eichstaedt CA, Song J, Benjamin N, Harutyunova S, Fischer C, Grunig E, Hinderhofer K. EIF2AK4 mutation as "second hit" in hereditary pulmonary arterial hypertension. Respir Res. 2016;17(1):141.
Castilho BA, Shanmugam R, Silva RC, Ramesh R, Himme BM, Sattlegger E. Keeping the eIF2 alpha kinase Gcn2 in check. Biochim Biophys Acta. 2014;1843(9):1948–68.
Nameki N, Yoneyama M, Koshiba S, Tochio N, Inoue M, Seki E, Matsuda T, Tomo Y, Harada T, Saito K, et al. Solution structure of the RWD domain of the mouse GCN2 protein. Protein Sci. 2004;13(8):2089–100.
Longchamp A, Mirabella T, Arduini A, MacArthur MR, Das A, Trevino-Villarreal JH, Hine C, Ben-Sahra I, Knudsen NH, Brace LE, et al. Amino acid restriction triggers angiogenesis via GCN2/ATF4 regulation of VEGF and H2S production. Cell. 2018;173(1):117–29 e114.
Chaveroux C, Lambert-Langlais S, Parry L, Carraro V, Jousse C, Maurin AC, Bruhat A, Marceau G, Sapin V, Averous J, et al. Identification of GCN2 as new redox regulator for oxidative stress prevention in vivo. Biochem Biophys Res Commun. 2011;415(1):120–4.
Yang H, Zeng Q, Ma Y, Liu B, Chen Q, Li W, Xiong C, Zhou Z. Genetic analyses in a cohort of 191 pulmonary arterial hypertension patients. Respir Res. 2018;19(1):87.
Acknowledgements
We thank all members of the studied families for their contribution to this research. Special thanks to Mrs. Inaam El-Rassy from the Molecular Core Facility at AUB for her technical assistance in Sanger Sequencing.
Funding
This work was supported by a grant from the Medical Practice Plan (MPP) and University Research Board (URB) grants from the American University of Beirut.
Author information
Authors and Affiliations
Contributions
WH and OA did the whole exome sequencing and interpretation of the data. HS, MA, FB, and IB recruited and clinically assessed the patients. GN and IB conceived the work, secured its funding and analyzed and interpreted the results. All authors contributed to the writing up of the manuscript and agreed to its content.All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study, data collection and genetic analysis were approved by the institutional review board (IRB) at the American University of Beirut Medical Center (AUBMC) (Protocol Number IM.IB.01). A written consent form was obtained from all participants in this study. Genetic analyses and return of genetic data were performed in accordance with protocols approved by the Partners Human Research Committee. The data include medical history, physical exam, family history for PAH, echocardiography, pulmonary function test, chest computerized tomography (CT), CT pulmonary angiography, ventilation perfusion scan and pro-Brain Natriuritic Peptide (BNP).
Consent for publication
Not applicable.
Competing interests
The authors report no competing interests. Those authors contributed equally to the work. Ossama K. Abou Hassan: oa20@aub.edu.lb, Wiam Haidar: wh30@aub.edu.lb, Mariam Arab: ma81@aub.edu.lb, Hadi Skouri: hs13@aub.edu.lb, Fadi Bitar: fbitar@aub.edu.lb, Georges Nemer: gn08@aub.edu.lb, Imad Bou Akl: ib08@aub.edu.lb.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Variant filtering workflow for exome sequencing of the patients. The left panel illustrates the type of unselected variants in the filter followed by exclusion of non-disease causing identified in polyphen, the right panel show the selected variants included in the filter followed by including all the allele frequencies < 10% to reach 50–70 identified causal variants associated with the disease. UTR = Untranslated Region; AF = allele frequency.
Additional file 2: Figure S2.
Predicted functional partners of the EIF2AK4 protein. https://string-db.org/cgi/network.pl?taskId=J7XBBrbsBLAi
Additional file 3: Figure S3.
Current and prior reported EIF2AK4 mutations detected in extensively worked up patients with PAH, PCH and PVOD diagnosis [5, 11, 12, 27]. The locations of the mutations are depicted on the rightmost and leftmost column and the consequence of the homozygous (blue) or compound mutations (red) and are defined as follows: The vertical black line depicts the protein structure for each patient reported, a point denotes a SNP mutation or stop codon differentiated by the COOH terminal continuation of the protein line structure, a solid red or blue line depicts a deletion, a diagonal line depicts a splice mutation, a dashed line denotes the affected/deleted haplo-insufficiency in relation to the compound chromosome mutation. The functional domains of the protein with respect to the mutation location is depicted in the middle
Additional file 4: Table S1.
List of Genes Linked to Pulmonary Arterial Hypertension.
Additional file 5: Table S2.
Variants in the coding regions of the EIF2AK1–4 genes in Individulas AII1 and AII.2 (excluding synonymous variants).
Additional file 6: Table S3.
Variants in the coding regions of the EIF2AK4 gene network in Individulas AII1 and AII.2 (excluding synonymous variants).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Abou Hassan, O.K., Haidar, W., Arabi, M. et al. Novel EIF2AK4 mutations in histologically proven pulmonary capillary hemangiomatosis and hereditary pulmonary arterial hypertension. BMC Med Genet 20, 176 (2019). https://doi.org/10.1186/s12881-019-0915-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-019-0915-7