
Abstract
Background
Wilson disease (OMIM # 277900) is a autosomal recessive disorder characterized by accumulation of copper in liver and brain. The accumulation of copper resulting in oxidative stress and eventually cell death. The disease has an onset in a childhood and result in a significant neurological impairment or require lifelong treatment. Another serious consequence of the disease is the development of liver damage and acute liver failure leading to liver transplant. The disorder is caused by mutations in the ATP7B gene, encoding a P-type copper transporting ATPase.
Case presentation
We performed genetic analysis of three unrelated patients from three different Vietnamese families. These patients had clinical features such as numbness of hands and feet, vomiting, insomnia, palsy, liver failure and Kayser–Fleischer (K–F) rings and were diagnosed with Wilson disease in the Human Genetics Department, Vietnam National Children’s Hospital. The entire coding region and adjacent splice sites of ATP7B gene were amplified and sequenced by Sanger method. Sequencing data were analyzed and compared with the ATP7B gene sequence published in Ensembl (ENSG00000123191) by using BioEdit software to detect mutations.
Conclusions
In this study, five mutations in the ATP7B gene were found. Among of these, three mutations were novel: c.750_751insG (p.His251Alafs*19) in exon 2, c.2604delC (p.Pro868Profs*5) in exon 11, and c.3077 T > A (p.Phe1026Tyr) in exon 14. Our results of the mutations associated with Wilson disease might facilitate the development of effective treatment plans.
Similar content being viewed by others


Background
Wilson disease (OMIM #277900) is characterized by an accumulation of copper in many organs and tissues [1]. Patients with Wilson disease typically present with low serum Cu and ceruloplasmin concentrations, increased urinary Cu excretion, and/or increased hepatic Cu concentrations, the presence of Kayser-Fleischer rings in the cornea, or neurological/psychiatric symptoms. There is a significantly higher frequency of Wilson disease in East Asian populations than in other populations, ranging from 1:1500 [2] to 1: 3000 [3]. However, clinical manifestations vary between individuals, even within families and in monozygotic twins [4,5,6]. There is also a wide range in the age of onset, including early-onset hepatic disease in a 3-year-old child and late-onset disease [1] with a mean age of onset of 15.9 years [7]. Indeed, failure diagnosis is the principle cause of death for patients with Wilson disease [8, 9].
Mutations in the ATP7B gene were identified as the cause of Wilson disease in 1993 and this gene is still the only one associated with the disorder [10,11,12]. ATP7B has an essential role in human physiology, particularly in liver and brain function. In the absence of ATP7B function, there is a toxic accumulation of copper in various body tissues, resulting in a wide variety of symptoms, including acute and chronic hepatitis, liver failure, and neurologic dysfunction. ATP7B maps on 13q14.3-q21, contains 21 exons and encodes a 1465-amino acids membrane protein [11,12,13]. ATP7B (copper-transporting ATPase) consists of six metal-binding domains, eight transmembrane segments, and an ATP-binding domain typical of copper ATPases with a P-domain, an N-domain, and an A-domain with the TGE sequence motif [14,15,16]. Wilson disease is typically caused by homozygous or compound heterozygous mutations in the ATP7B gene. Over 776 mutations have been detected in the ATP7B gene until now (The Human Gene Mutation Database. http://www.hgmd.cf.ac.uk/ac/index.php). Wilson disease invariably results in severe disability and death, especially for patients with no apparent clinical manifestations who are untreated. The identification of mutations in the ATP7B gene is the one of the useful diagnostic tools for diagnosis and treatment orientation for patients with Wilson disease.
In this study, we identified mutations in the ATP7B gene of three patients’ families with Wilson disease by sequencing the entire coding region and adjacent splice sites of the ATP7B gene. Information about mutations in the ATP7B gene will be helpful for efficiently diagnosing Wilson disease and providing early therapeutic intervention for patients.
Case presentation
Clinical presentation
Patient 1 (WBW140801)
He was an 8-year-old boy who was hospitalized with clinical features such as numbness of hands and feet, vomiting, insomnia, palsy and Kayser–Fleischer (K–F) rings. Biochemical indices of the blood serum revealed 0.0032 mg/dL serum ceruloplasmin (normal is 20–35 mg/dL), 49 μg/dL serum free copper (normal is < 15 μg/dL), 20.1 IU/L ALT (alanine aminotransferase) (normal is < 40 IU/L), 26.82 IU/L AST (aspartate aminotransferase) (normal is < 40 IU/L), and PT (prothrombin time) of 49% (normal is > 70%). In addition, biochemical indices of the urine revealed 580 μg/24 h urinary copper (normal is < 60–100 μg/24 h). The patient had liver failure and severe neurological symptoms. He was diagnosed with Wilson disease in the Human Genetics Department, Vietnam National Children’s Hospital. His parents and his half brother had a normal phenotype. However, we did not collect a sample from his father for genetic analysis.
Patient 2 (WBW100604)
She was an 8-year-old girl who was hospitalized with clinical features such as oedema, elevated serum transaminase, liver failure, and Kayser–Fleischer rings. Biochemical indices of the blood serum revealed 0.019 mg/dL serum ceruloplasmin (normal is 20–35 mg/dL), 116.9 IU/L ALT (alanine aminotransferase) (normal is < 40 IU/L), 139.1 IU/L AST (aspartate aminotransferase) (normal is < 40 IU/L), and PT (prothrombin time) of 22% (normal is > 70%). In addition, biochemical indices of the urine revealed 150 μg/24 h urinary copper (normal is < 60–100 μg/24 h). She was diagnosed with Wilson disease in the Human Genetics Department, Vietnam National Children’s Hospital. Her parents and her younger sister had a normal phenotype.
Patient 3 (WBW170704)
He was a 10-year-old boy who was hospitalized with clinical features such as oedema, jaundice, acute liver failure, and Kayser–Fleischer rings. Biochemical indices of the blood serum revealed 0.092 mg/dL serum ceruloplasmin (normal is 20–35 mg/dL), 11.6 μg/dL serum free copper (normal is < 15 μg/dL), 55.7 IU/L ALT (alanine aminotransferase) (normal is < 40 IU/L), 129.0 IU/L AST (aspartate aminotransferase) (normal is < 40 IU/L), and PT (prothrombin time) of 17% (normal is > 70%). In addition, biochemical indices of the urine revealed 3639 μg/24 h urinary copper (normal is < 60–100 μg/24 h). He was diagnosed with Wilson disease in the Human Genetics Department, Vietnam National Children’s Hospital. His parents and his younger sister had a normal phenotype.
Genetic analysis
Genomic DNA was isolated from peripheral blood samples (including sample from patients and their families) using a Qiagen DNA blood mini kit (QIAamp DNA Blood Mini preparation kits, German) following the manufacturer’s guidelines. The DNA concentration was determined using a Thermo Scientific NanoDrop spectrophotometer (Waltham, MA, USA).
All 21 exons and exon-intron boundaries of the ATP7B gene were amplified and analysed by direct sequencing. Oligonucleotide primers were synthesized and purchased from IDT (USA) (Additional file 1: Table S1). Fifty nanograms of genomic DNA was subjected to 35 cycles of PCR amplification in a 25 μL volume consisting of 10X PCR buffer (Invitrogen, USA), 10 μM concentration of each primer, 20 mM MgCl2, 10 μM dNTPs, and 5 U Taq DNA polymerase (Invitrogen, USA). DNA was denatured at 95 °C for 12 min followed by 35 cycles of denaturation for 1 min at 95 °C, annealing for 1 min at 60–65 °C, and extension for 1 min at 72 °C, and a final extension for 7 min at 72 °C. PCR amplification was carried out on an ABI 9700 GeneAmp PCR system (USA).
DNA sequencing was performed in both directions, initiated from forward and reverse primers, which had been used in an initial PCR reaction. PCR products were purified with Qiagen Purification kit (QIAquick PCR Purification Kit, Germany) and sequenced on ABI PRISM 3130 Genetic Analyser machine (USA). Sequencing data were analysed by using SeqScape 2.5 software, Chromas or equivalent software and compared with the ATP7B gene sequence published in Ensembl (ENSG00000123191) by using BioEdit software to detect mutations.
In silico analysis tools
The consequence of any novel nonsynonomous nucleotide variations that were identified within exons were evaluated with the in silico analysis tools sorting intolerant from tolerant (SIFT) prediction [17], polyphen 2 [18] and mutation taster [19].
Discussion and conclusions
Patient 1
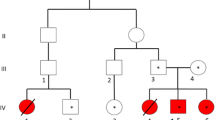
A heterozygous mutation in intron 12 (c.2866-2A > G) and a heterozygous mutation in exon 14 (p.Phe1026Tyr) of the ATP7B gene were identified in patient 1. Genetic analysis in his mother showed that she had a heterozygous mutation in exon 14 (p.Phe1026Tyr) but did not have the mutation in intron 12. This result was found the in genotype of the patient’s half brother (Fig. 1). The impact possibility of a missense mutation was evaluated by using bioinformatics tools such as SIFT [17], PolyPhen2 [18] and Mutation Taster [19]. Polyphen-2 analysis for p.Phe1026Tyr substitution in the ATP7B protein of patient 1 indicated a “probably damaging” status with a score of 1.0. Mutation Taster tool predicted the mutation to be “disease causing” with a score of 0.99, and analysis performed by SIFT resulted in a score of 0.01, which indicated that the substitution is “damaging”. These mutations could be the cause of his severe symptoms and early onset of disease. The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php), and Wilson Disease Mutation Database (http://www.wilsondisease.med.ualberta.ca/search3.asp) were used to determine that the p.Phe1026Tyr change was novel. In addition, c.2866-2A > G was a known mutation with code number CS136099 in The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) and its gnomAD frequency was 0.012190% (Table 1).

Mutations of the ATP7B gene were identified by sequencing in patient 1 and his family. Pedigree of patient’s family (a), the Kayser-Fleischer ring around the cornea of patient’s eye (b) and mutations (c) of ATP7B gene were identified, including a compound heterozygous mutation p.Phe1026Tyr and heterozygous mutation c.2866-2A > G
Genetic analysis showed that patient 1 had a p.Phe1026Tyr mutation, which was located in exon 14 and the phosphorylation domain (P-domain) of the ATP7B protein. In addition, the patient also carried a mutation in intron 12 (c.2866-2A > G). Patient 1 had the very typically symptoms of the disease such as numbness of hands and feet, vomiting, insomnia, palsy and Kayser–Fleischer rings. Blood and urine biochemical tests manifested that the index of serum ceruloplasmin was very low at 0.0032 mg/dL (Table 2), whereas the concentration of free copper in serum and 24 h urine sample were 3- to 5- fold higher than normal, respectively (serum and urine copper levels of 49 μg/dL and 580 μg, respectively). In addition, a drop in the PT index to 49% indicated a dramatic decline in liver function. The results of in genetic analysis and biochemical tests clearly provided evidence of genotypic and phenotypic correlations in the patient.
Patient 1 had a very severe and characteristic phenotype of Wilson disease (with clinical features such as numbness of hands and feet, vomiting, insomnia, palsy, liver failure and Kayser–Fleischer rings). This result could be explained by the fact that the patient carried two mutations that were severely affecting the function of protein ATP7B. The mutation p.Phe1026Tyr was located in exon 14 and the phosphorylation domain (P-domain) of the ATP7B protein, which was supposed to have a significant effect on normal protein function. Forbes et al. [20] has given evidence for the effects of ATP7B mutations in severe neuropsychiatric deterioration. Ljubic et al. [21] indicated that mutations in the P-domain interfered with catalytic phosphorylation. In addition, this mutation was compounded by the heterozygous mutation c.2866-2A > G located in the splice site, which can affect mRNA splicing [22].
Patient 2
Two heterozygous mutations in exon 2 (p.Ser105* and p.His251Alafs*19) of the ATP7B gene were identified in patient 2. In these mutations, c.314C > A (p.Ser105*) (with gnomAD frequency of 0.008128%) was a known mutation and c.750_751insG (p.His251Alafs*19) (with a gnomAD frequency of 0.004730%) was a novel mutation. Genetic analysis of her family exposed that her father had the heterozygous mutation p.Ser105* and her mother had the heterozygous mutation p.His251Alafs*19 (Fig. 2).

Mutations of the ATP7B gene were identified by sequencing in patient 2 and his family. Pedigree of patient’s family (a), the Kayser-Fleischer ring around the cornea of patient’s eye (b) and mutations (c) of ATP7B gene were identified, including a compound heterozygous mutation p.Ser105* and heterozygous frameshift mutation p.His251Alafs*19
Patient 2 carried two heterozygous mutations, p.Ser105* and p.His251Alafs*19, in exon 2, leading to complete loss of the protein function and early-onset disease. The study by Huong et al. [23] on Vietnamese patients showed that mutations in exon 2 and the p.Ser105* mutation are quite common at a high rate (39.6 and 32.6%, respectively). Exon 2 is the hotspot region of the ATP7B gene in many populations, such as Chinese Indian populations, and mutations in exons 2–5 associated with severe phenotypes have been found in the Indian population [24,25,26]. The investigate of Chen et al. [27] revealed that missense or nonsense mutations caused by single nucleotide variant were very popular (60%), followed by insertions/deletions (26%) and splice-site mutations (9%) in the genetics of Wilson disease. It has previously been suggested that age of onset could potentially be related to the functional activity of ATP7B mutants and that truncating mutations in the ATP7B gene were associated with an early onset of Wilson disease. Gromadzka et al. [28] noticed that the age of onset was 14 ± 7 years in patients with two severe truncating mutations. In addition, similar results were found by Merle et al. [29], who reported that the age of onset of these patients was in the range of 9–13 years.
Patient 3
In patient 3, a novel heterozygous mutation c.2604delC (p.Pro868Profs*5) was detected in exon 11 of the ATP7B gene. Patient 3 also had a heterozygous mutation p.Ser105*. Genetic analysis has shown that the heterozygous mutation p.Ser105* was inherited from his father and that the heterozygous mutation p.Pro868Profs*5 was inherited from his mother (Fig. 3).

Mutations of the ATP7B gene were identified by sequencing in patient 3 and his family. Pedigree of patient’s family (a), the Kayser-Fleischer ring around the cornea of patient’s eye (b) and mutations (c) of ATP7B gene were identified, including a compound heterozygous mutation p.Ser105* and heterozygous frameshift mutation p.Pro868Profs*5
Patient 3 carried two severe truncating mutations, which is similar to patient 2. In addition, both patients (patients 2 and 3) had liver damage with early onset. Kalach et al. [30] and Wilson et al. [31] reported cirrhosis in a 3-year-old patient and acute liver failure in a 5-year-old patient, respectively. Yi et al. [32], who established neural and hepatic differentiation platforms of patient-derived induced pluripotent stem cells, showed that liver and brain are the most affected organs. Previous studies have shown that a severe impairment of copper transport resulting in severe liver damage usually occurs in the early stages of Wilson disease [1, 33, 34]. There are rare patients, who present with neurological disease but do not have liver cirrhosis [1].
In both patients 2 and 3, who had the combination of a p.Ser105* mutation and a p.His251Alafs*19 or p.Pro868Profs*5 mutation, the liver was severely damaged with high expression levels of liver enzymes, such as aminotransferases (Table 2). The patients had a high level of aspartate aminotransferase (AST) (139.1 IU/L in patient 2 and 129.0 IU/L in patient 3) and alanine aminotransferase (ALT) (116.9 IU/L in patient 2 and 55.7 IU/L in patient 3). These enzymes are normally predominantly contained within liver cells so that if the liver is injured or damaged, the liver cells spill these enzymes into the blood, raising the AST and ALT enzyme blood levels and signalling liver disease. In these patients, minor reductions in serum ceruloplasmin (with 0.0190 mg/dL and 0.0920 mg/dL, respectively, in patient 2 and 3), serum free copper and the PT index (with 22 and 17%, respectively, for patient 2 and 3) have been observed. Increased urine copper excretion was found in these patients with 150 mg/24 h urine (patient 2) and 3639 mg/24 h urine (patient 3). This fiding is a typical symptom of Wilson disease due to decreased serum binding of copper to ceruloplasmin.
In our study, we identified five nucleotide changes in the ATP7B gene in three patients with Wilson disease from three unrelated Vietnamese families. Three of these changes were novel mutations: p.His251Alafs*19 in exon 2, p.Pro868Profs*5 in exon 11, and p.Phe1026Tyr in exon 14. These results provide knowledge on ATP7B mutations in patients with Wilson disease and might contribute to the development of effective treatment plans for these patients.
Abbreviations
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- ATP7B:
-
ATPase copper transporting beta
- DNA:
-
Deoxyribonucleic acid
- mRNA:
-
Messenger RNA
- PCR:
-
Polymerase chain reaction
- PT:
-
Prothromlin time
- RNA:
-
Ribonucleic acid
- SIFT:
-
Sorting intolerant from tolerant
References
Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson's disease. Lancet. 2007;369:397–408.
Ohura T, Abukawa D, Shiraishi H, Yamaguchi A, Arashima S, Hiyamuta S, Tada K, Iinuma K. Pilot study of screening for Wilson disease using dried blood spots obtained from children seen at outpatient clinics. J Inherit Metab Dis. 1999;22:74–80.
Hahn SH, Lee SY, Jang YJ, Kim SN, Shin HC, Park SY, Han HS, Yu ES, Yoo HW, Lee JS, Chung CS, Lee SY, Lee DH. Pilot study of mass screening for Wilson’s disease in Korea. Mol Genet Metab. 2002;76:133–6.
Czlonkowska A, Rodo M, Gromadzka G. Late onset Wilson’s disease: therapeutic implications. Mov Disord. 2008;23:896–8.
Czlonkowska A, Gromadzka G, Chabik G. Monozygotic female twins discordant for phenotype of Wilson’s disease. Mov Disord. 2009;24:1066–9.
Kegley KM, Sellers MA, Ferber MJ, Johnson MW, Joelson DW, Shrestha R. Fulminant Wilson’s disease requiring liver transplantation in one monozygotic twin despite identical genetic mutation. Am J Transplant. 2010;10:1325–9.
Taly AB, Meenakshi-Sundaram S, Sinha S, Swamy HS, Arunodaya GR. Wilson disease: description of 282 patients evaluated over 3 decades. Medicine (Baltimore). 2007;86:112–21.
Czlonkowska A, Tarnacka B, Litwin T, Gajda J, Rodo M. Wilson’s disease-cause of mortality in 164 patients during 1992-2003 observation period. J Neurol. 2005;252:698–703.
Walshe JM. Cause of death in Wilson disease. Mov Disord. 2007;22:2216–20.
Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5(4):327–37.
Petrukhin K, Fischer SG, Pirastu M, Tanzi RE, Chernov I, Devoto M, Brzustowicz LM, Cayanis E, Vitale E, Russo JJ, et al. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet. 1993;5(4):338–43.
Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, Romano DM, Parano E, Pavone L, Brzustowicz LM, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5(4):344–50.
White A, Tomfohrde J, Stewart E, Barnes R, Le Paslier D, Weissenbach J, Cavalli-Sforza L, Farrer L, Bowcock A. A 4.5-megabase yeast artificial chromosome contig from human chromosome 13q14.3 ordering 9 polymorphic microsatellites (22 sequence-tagged sites) tightly linked to the Wilson disease locus. Proc Natl Acad Sci U S A. 1993;90(21):10105–9.
Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87(3):1011–46.
Lutsenko S, LeShane ES, Shinde U. Biochemical basis of regulation of human copper-transporting ATPases. Arch Biochem Biophys. 2007;463(2):134–48.
Bartee MY, Lutsenko S. Hepatic copper-transporting ATPase ATP7B: function and inactivation at the molecular and cellular level. Biometals. 2007;20(3–4):627–37.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–82.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Schwarz JM, Rӧdelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–6.
Forbes N, Goodwin S, Woodward K, Morgan DG, Brady L, Coulthart MB, Tarnopolsky MA. Evidence for synergistic effects of PRNP and ATP7B mutations in severe neuropsychiatric deterioration. BMC Med Genet. 2014;15(22). https://doi.org/10.1186/1471-2350-15-22.
Ljubić H, Kalauz M, Telarović S, Ferenci P, Ostojić R, Noli MC, Lepori MB, Hrstić I, Vuković J, Premužić M, Radić D, Ravić KG, Sertić J, Merkler A, Barišić AA, Loudianos G, Vucelić B. ATP7B gene mutations in Croatian patients with Wilson disease. Genet Test Mol Biomarkers. 2016;20(3):112–7.
Aggarwal A, Chandhok G, Todorov T, Parekh S, Tilve S, Zibert A, Bhatt M, Schmidt HHJ. Wilson disease mutation pattern with genotype-phenotype correlations from western India: confirmation of p.C271* as a common Indian mutation and identification of 14 novel mutations. Ann Hum Genet. 2013;77:299–307.
Huong MTN, Hoa APN, Mai PTN, Ngoc DN, Van TT, Hai TL, Chi VP. Mutation analysis of the ATP7B gene in 43 Vietnamese Wilson’s disease patients. World Acad Sci Eng Technol. 2017;11(2):174–179.
Chuang LM, Wu HP, Jang MH, Wang TR, Sue WC, Lin BJ, Cox DW, Tai TY. High frequency of two mutations in codon 778 in exon 8 of the ATP7B gene in Taiwanese families with Wilson disease. J Med Genet. 1996;33:521–3.
Payne AS, Kelly EJ, Gitlin JD. Functional expression of the Wilson disease protein reveals mislocalization and impaired copper-dependent trafficking of the common H1069Q mutation. Proc Natl Acad Sci U S A. 1998;95:10854–9.
Wu ZY, Wang N, Lin MT, Fang L, Murong SX, Yu L. Mutation analysis and the correlation between genotype and phenotype of Arg778Leu mutation in Chinese patients with Wilson disease. Arch Neurol. 2001;58:971–6.
Chen C, Shen B, Xiao JJ, Wu R, Canning SJD, Wang XP. Currently clinical views on genetics of Wilson’s disease. Chinese Med J. 2015;128(13):1826–30.
Gromadzka G, Schmidt HH, Genschel J, Bochowd B, Rodoe M, Tarnacka B, Litwin T, Chabik G, Członkowsk A. Frameshift and nonsense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson’s disease. Clin Genet. 2005;68:524–32.
Merle U, Weiss KH, Eisenbach C, Tuma S, Ferenci P, Stremmel W. Truncating mutations in the Wilson disease gene ATP7B are associated with very low serum ceruloplasmin oxidase activity and an early onset of Wilson disease. BMC Gastroenterol. 2010;10(8). https://doi.org/10.1186/1471-230X-10-8
Kalach N, Seidman EG, Morin C, Rasquin-Weber A, O’Regan S, Laberge JM, Blanchard H, Russo P, Paradis K. Acute liver failure from Wilson’s disease in a five year-old child. Can J Gastroenterol. 1993;7:610–2.
Wilson DC, Phillips MJ, Cox DW, Roberts EA. Severe hepatic Wilson’s disease in preschool-aged children. J Pediatr. 2000;137:719–22.
Yi F, Qu J, Li M, Suzuki K, Kim NY, Liu GH, Belmonte JC. Establishment of hepatic and neural differentiation platforms of Wilson’s disease specific induced pluripotent stem cells. Protein Cell. 2012;3(11):855–63.
Ferenci P. Wilson’s disease. Clin J Gastroenterol Hepatol. 2005;3(8):726–33.
Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089–111.
Acknowledgements
The authors appreciate the participation of the patient’s family in this study. This work was supported by Institute of Genome Research, Vietnam Academy of Science and Technology and Vietnam National Children’s Hospital, Ministry of Health. We would like to thank Prof. Han Wook Yoo in Genome Research Center for Birth Defects and Genetic Disease, Asian Institute for Life Sciences, Asian Medical Center, Seoul, Korea.
Funding
This study was supported by Institute of Genome Research, Vietnam Academy of Science and Technology and Vietnam National Children’s Hospital, Ministry of Health.
Availability of data and materials
Not applicable. We are submitting the data together with this manuscript.
Author information
Authors and Affiliations
Contributions
NTMH conceived and coordinated the study, analyzed data. NTKL contributed to data analysis and wrote the manuscript. NDN, NTPM, NPAH prepared the samples, performed experiments and help to draft the manuscript. LTH, PVC, TTV, TVK, NHH contributed to study design and help to manuscript preparation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Scientific Committee of Institute of Genome Research under reference number 12/QD-NCHG.
Consent for publication
Written informed consent for the publication of the patient’s medical data was obtained from the parents before collecting samples. A copy of the written consent is available for review by the editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. List of PCR primers. (DOCX 15.6 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Huong, N.T.M., Lien, N.T.K., Ngoc, N.D. et al. Three novel mutations in the ATP7B gene of unrelated Vietnamese patients with Wilson disease. BMC Med Genet 19, 104 (2018). https://doi.org/10.1186/s12881-018-0619-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0619-4