
Abstract
Background
Tricho-rhino-phalangeal syndrome (TRPS) is an autosomal dominant disorder characterized by craniofacial and skeletal malformations including short stature, thin scalp hair, sparse lateral eyebrows, pear-shaped nose and cone shaped epiphyses. This condition is caused by haploinsufficiency of the TRPS1 gene. Previous genotype-phenotype studies have correlated exon 6 missense mutations with TRPS type III, a severe form of type I with pronounced, facial characteristics, short stature and brachydactyly and differing from type II by the absence of exostoses and mental retardation.
Case presentation
We report the first case of a Moroccan family, a father and his three children, in which the diagnosis of type III TRPS was suspected based on severe clinical and radiological features. Molecular analysis of the TRPS1 gene revealed a novel missense mutation in exon 6, (p.Ala932Ser), located in the GATA-type DNA-binding zinc finger domain.
Conclusion
Our observations in this kindred support the previous genotype-phenotype results suggesting that patients with more pronounced facial characteristics and more severe shortening of hands and feet are more likely to have mutation in exon 6 of TRPS1.
Similar content being viewed by others


Background
Tricho-rhino-phalangeal syndrome (TRPS) is a malformation syndrome characterized by short stature, sparse and slowly growing hair, a distinctive facies with rarefaction of lateral eyebrows, bulbous nasal tip, a high philtrum and thin upper lip. In addition, patients present cone-shaped epiphyses and severe generalized shortening of all phalanges, metacarpals and metatarsal bones [1]. This syndrome was first described by Giedion in 1966 [1]. Since then more than 120 patients have been described in the literature. Three subtypes of TRPS sharing all the common distinctive features have been described; type I which is the classical form of TRPS. Type II also called Langer-Giedion syndrome, which is distinguished from type I by the presence of multiple bony or cartilaginous exostoses in addition to mild mental retardation. This condition represents a contiguous gene syndrome on chromosome 8q24 [2]. Type III or Sugio-Kanjii Syndrome is characterized by severe shortness of all metacarpals, phalanges and stature without exostoses or mental retardation [3].
TRPS is usually inherited in an autosomal dominant manner [4], and caused by pathogenic mutations in the TRPS1 gene (OMIM 604386), located on chromosome 8q23.3 and encoding a zinc finger transcriptional repressor with 2 potential nuclear localization signals and 9 different zinc-finger motifs [5]. The TRPS1 protein appears to be involved in the regulation of the development of chondrocyte and perichondrium [6, 7]. Nonsense mutations are mostly found in patients with the TRPS I phenotype, while TRPS II is a contiguous gene deletion syndrome involving loss of the TRPS1 and the EXT1 genes, the latter being mutated in multiple exostosis type I. Patients with the more severe phenotype, described as TRPS type III, have been found to carry missense mutations in the GATA-type DNA-binding zinc finger domain encompassing codons 903 to 953 [6]. Recently, genotype-phenotype studies showed that TRPS1 exon 6 mutations, encompassing codons 902 to 941, may result in more pronounced facial characteristics and more severe shortening of hands and feet compared to mutations located in other TRPS1 exons, but cohort numbers are still scarce to allow firm conclusions [8]. Several authors reported the management of TRPS short stature by growth hormone (GH) supplementation [9,10,11]. The results available to date are, however controversial.
Here, we report the first case of a Northern African family of 3 siblings and their father with a TRPS type III phenotype. The identification of a novel missense mutation, (p.Ala932Ser), in TRPS1 exon 6 in this kindred provides further support that mutations in this exon may be correlated with a more pronounced features of the syndrome.
Case presentation
We describe here a TRPS family with three affected siblings from a consanguineous marriage and their affected father (Fig. 1).

Pedigree of the family. The arrow indicates patient 1 which is the proband
Patient 1
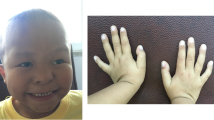
A 13 years old girl was referred to the department of medical genetics in Rabat for short stature and brachydactyly. No prenatal investigations had been performed and the motor development was normal. Clinical examination found a proportionate short stature ≥ 3 SD (139 cm), sparse thin and hypo pigmented hair, thick eyebrows with lateral rarefaction, a characteristic pear-shaped nose, a long philtrum and a thin upper lip (Fig. 2a). No intellectual impairment was noticed. Extremities examination showed brachydactyly with bilateral axial deviation of third and fourth fingers, a short fifth metacarpal on the left, flat feet with partial syndactyly of the second and third toes on the right (Fig. 3a). Radiological examinations disclosed flattened femoral heads, poorly covered with a short and widened femoral neck predominantly on the right side in the pelvis radiograph. Hands’ radiographs showed misalignment of the middle and distal phalanges prevailing at the third and fourth fingers with irregular appearance of inverted V of the middle phalanges with cone-shaped corresponding epiphysis and a shortness of the fifth left metacarpal. Radiography of the feet found a bilateral misalignment of the distal phalanx of the hallux with a widened appearance of inverted V of the middle phalanges with cone shaped corresponding epiphysis. Spine and limbs radiographs in addition to the patient’s karyotype were all normal.

Typical dysmorphological features of TRPS III in the family members showing thick eyebrows with lateral rarefaction, characteristic pear-shaped nose, long philtrum and thin upper lip. a- Proband, b- Sister, c- Brother, d- Father

Characteristic hands and feet disclosing severe brachydactyly with syndactyly in the family members. a- Proband, b- Sister, c- Brother, d- Father. X-rays of the proband show misalignment of the middle and distal phalanges predominant at the third and fourth fingers with cone shaped epiphyses in hands and feet
Patient 2
The older sister of the proband was 18 years old; she presented a proportionate short stature (142 cm ≥ 3 SD) with thin, sparse and slow growing hair and the same distinctive facies characteristics as her sister, thick eyebrows and bulbous nasal tip (Fig. 2b). She showed also a brachydactyly with clinodactyly, deviation of the middle fingers on both hands, and partial syndactyly of the second and third toes on both feet (Fig. 3b). She had no intellectual impairment and no radiological exams were performed.
Patient 3
The elder brother of the siblings was 21 years old; with no mental disorder, he presented a proportionate short stature (148 cm, ≥ 3 SD), thin scalp hair, rarefaction of lateral eyebrows and a typical pear-shaped nose with a long philtrum (Fig. 2c). He presented brachydactyly in both hands and feet with shortness of the right middle finger (Fig. 3c). The patient declined radiological exam of extremities.
Patient 4
The father of patients 1, 2 and 3 presented with a severe proportionate short stature (141 cm, ≥ 3 SD), diffuse alopecia with fine hair, absence of lateral eyebrows, large beaked nose and a long philtrum with thin upper lip (Fig. 2d). He had short metacarpals with brachydactyly, axial deviation of the middle finger and racket nails (Fig. 3d). No radiological investigations were performed.
Mutation analysis
Method: Following genetic counselling informed consent was obtained from all family members for both, molecular analysis and publication of clinical photographs. DNA was extracted from the three siblings and their two parents from EDTA blood samples using the QIAamp DNA Blood Mini Kit (Qiagen) according to the manufacturer’s protocol. Analysis of the entire coding region (exons 2 to 7, including intron/exon-boundaries) of the TRPS1 gene (RefSeq: NM_014112.4) was performed by PCR amplification and bi-directional Sanger sequencing as previously described [12]. Gene dosage analysis was performed using the MLPA assay (P228, Lot No.: 0514, MRC Holland, NL). In silico prediction analysis was performed using SIFT (sift.jcvi.org) [13], MutationTaster (MT) [14] and PolyPhen-2 (PP2) [15] programs.
In TRPS1 exon 6 a heterozygous germline sequence variant, c.2794G > T, was identified in patient 1 (Fig. 4). Subsequent targeted mutation analysis of exon 6 of the remaining family members confirmed segregation of the sequence variant in the affected father (Case 4), and the two affected siblings (Cases 2 and 3). The healthy mother did not carry the sequence variant. This missense mutation leads to an amino acid change at codon 932, (p.Ala932Ser), replacing the nonpolar residue alanine by a polar one, serine. In silico prediction programs rat this mutation, occurring at an evolutionary highly conserved amino acid residue, as “disease causing” (MT), “damaging” (SIFT) and “probably damaging” (PP2, HumVar score: 0.999). Furthermore, two different missense mutations located at amino acid residue 932, (p.Ala932Thr) and (p.Ala932Val), have already been described as pathogenic in 4 index patients [6, 8, 16]. According to the ACMG criteria for classifying pathogenic variants [17], the (p.Ala932Ser) variant is classified as “likely pathogenic” (PM1, PM2, PM5, PP1, PP2, 149 PP3, PP4). This variant was absent in an in-house database of 770 exomes, and absent in the publicly available databases, including the Exome Aggregation Consortium [18], ExomeVariantServer [19] and dbSNP [20].

Electrophoregram showing above the wild-type sequence, and below the heterozygous sequence variant c.2794G > T identified in exon 6 of the TRPS1 gene
Discussion
In 1966, Giedion reported several individuals with common manifestations including growth impairment, sparse hair, unusual face with characteristic shape of the nose and anomalies of the distal limbs, he named this entity tricho-rhino-phalangeal syndrome (TRPS) [1]. Three years after this description, Langer et al and Gorlin et al described patients with the same entity associated with multiple exostoses and developmental delay [21, 22]. Hall et al. suggested subdividing TRPS into two subtypes: TRPS I (OMIM 190350) for patients with normal development and without exostoses, and TRPS II or Langer-Giedion syndrome (OMIM 150230) for individuals with intellectual disorder and exostoses.
After this subdivision, several authors have proposed another subtype, TRPS III (OMIM 190351). This condition is associated with more marked growth delay and severe generalized shortening of all phalanges and metacarpals [3, 6, 23, 24]. Some authors discussed that TRPS III could be considered as part of TRPS I entity with more severe manifestation based on the small phenotypic differences and intrafamilial variability [6, 8, 23]. In our family, however, there was no evident intrafamilial clinical variability; growth delay ranged between ≥ 2.5 and ≥ 3 SD and severe brachydactyly in hands and feet was evident in all family members. Additional associated physical signs reported in TRPS type I, such as endocrine disturbance was not present in either of our family members [8, 25,26,27] In 2000, Momeni et al. reported TRPS1 as the causal gene of TRPS [5]. This gene maps to 8q23.3 and encodes for a 141 kD multi zinc-finger transcription factor protein composed of 1294 amino acids with 9 putative zinc-finger motifs. The seventh zinc- finger motif binds to the GATA consensus cis element. TRPS1 is assumed to be a nuclear regulator of chondrocyte proliferation and differentiation and hair follicles proliferation by homodimerization of the complex with GATA binding protein sequences in DNA to repress transcription of target genes [9, 28]. Thus TRPS1 haploinsufficiency impairs endochondral cartilage differentiation and epithelial cell interactions in developing hair follicles by altering many signaling pathways as STAT3 and WNT/β catenin [7, 28,29,30]. Many studies reported mutations in the TRPS1 gene and to date it appears to be the only gene associated with the TRPS phenotype [6, 8, 9, 27, 28]. According to the Human Genome Mutation Database, more than 120 pathogenic mutations correlated to TRPS phenotype have been reported, with most of them being nonsense or frameshift mutations. Genotype-phenotype studies showed that missense mutations associated to TRPS exclusively occurred in exons 6 and 7, with most of them being located in exon 6 which encodes a GATA-type DNA-binding zinc finger domain [8]. Furthermore, patients with missense mutations in exon 6 seem to have more pronounced facial characteristics and shortening of hands and feet compared to mutations located in other exons [6, 8]. Still, data on more TRPS families and patients are needed to allow firm conclusions. In our family a novel missense mutation in exon 6, (p.Ala932Ser), was clearly associated with a TRPS type III phenotype in all family members. The mutation is considered to be likely pathogenic since it involves an evolutionary highly conserved amino acid residue, is absent from controls (Exome Sequencing Project, 1000 Genomes Project), co-segregates with the phenotype and different missense changes at codon 932 have already been reported as pathogenic, however we could not deduce a clear overlap between the phenotype of our family and that of other patients carrying mutations in the same codon [6, 8]. The (p.Ala932Ser) mutation probably results in decreased affinity to DNA and thus may exert a dominant negative effect as a component of a multimeric protein complex [6, 16]. TRPS is a condition inherited in an autosomal dominant manner [4]. Approximately, half of mutations reported in the literature were inherited while the other half represents de novo mutations [31, 32]. In our family, it was evident that the affected father had transmitted the missense mutation to his 3 children in autosomal dominant manner. However some authors reported the occurrence of the disease in siblings from healthy parents suggesting an autosomal recessive transmission [33, 34]. Corsini et al. reported on a patient with a heterozygous mutation in the TRPS1 gene whose healthy mothers was carrier of the mutation in the form of somatic mosaicism estimated at 10% in maternal blood-derived DNA using next generation sequencing. He proposed then a 5–10% risk of recurrence of TRPS1 for healthy parents with an affected child, in all patients with presumed de novo mutations, based on the known recurrence risk in other dominant conditions with demonstrated germinal mosaicism, such as Cornelia de Lange syndrome and Rubinstein-Taybi syndrome [35].
Conclusions
We report here the first case of a familial TRPS type III in Morocco with a novel TRPS1 missense mutation, (p.Ala932Ser), associated with a more pronounced dysmorphic features and further supporting the phenotype-genotype studies suggesting an association between TRPS type III and missense mutations in TRPS1 exon 6.
Abbreviations
- ACMG:
-
Annual clinical genetics meeting
- DNA:
-
Deoxyribonucleic acid
- EDTA:
-
Ethylene-diamine-teraacetic acid
- EXT:
-
Exostosin
- GH:
-
Growth Hormone
- IGF-1:
-
Insulin-like growth factor 1
- MLPA:
-
Multiplex ligation-dependent probe amplification
- MT:
-
Mutation Taster
- OMIM:
-
Online Mendelian Inheritance in Man
- PCR:
-
Polymerase chain reaction
- SD:
-
Standard deviation
- SIFT:
-
Sorting intolerant from tolerant
- STAT:
-
Signal Transducer and Activator of Transcription
- TRPS:
-
Tricho-rhino-phalangeal syndrome.
- WNT:
-
Wingless integration site
References
Giedion A. Tricho-rhino-phalangeal syndrome [Article in German]. Helv Paediatr Acta. 1966;21:475–85.
Nardmann J, Tranebjaerg L, Horsthemke B, Lüdecke HJ. The tricho-rhino-phalangeal syndromes: frequency and parental origin of 8q deletions. Hum Genet. 1997;99:638–43.
Niikawa N, Kamei T. The Sugio-Kajii syndrome, proposed tricho-rhino-phalangeal syndrome type III. Am J Med Genet. 1986;24:759–60.
Giedion A, Burdea M, Fruchter Z, Meloni T, Trosc V. Autosomal-dominant transmission of the tricho-rhino-phalangeal syndrome. Report of 4 unrelated families, review of 60 cases. Helv Paediatr Acta. 1973;28:249–59.
Momeni P, Glöckner G, Schmidt O, von Holtum D, Albrecht B, Gillessen-Kaesbach G, et al. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino- phalangeal syndrome type I. Nat Genet. 2000;24:71–4.
Lüdecke HJ, Schaper J, Meinecke P, Momeni P, Gross S, von Holtum, et al. Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet. 2001;68:81–91.
Napierala D, Sam K, Morello R, Zheng Q, Munivez E, Shivdasani RA, Lee B. Uncoupling of chondrocyte differentiation and perichondrial mineralization underlies the skeletal dysplasia in tricho-rhino-phalangeal syndrome. Hum Mol Genet. 2008;17:2244–54.
Maas SM, Shaw AC, Bikker H, Lüdecke HJ, van der Tuin K, Badura-Stronka M, Hennekam RC. Phenotype and genotype in 103 patients with tricho-rhino-phalangeal syndrome. Eur J Med Genet. 2015;58(5):279–92.
Merjaneh L, Parks JS, Muir AB, Fadoju D. A novel TRPS1 gene mutation causing trichorhinophalangeal syndrome with growth hormone responsive short stature: a case report and review of the literature. Int J Pediatr Endocrinol. 2014;2014(1):16.
Sohn YB, Ki CS, Park SW, Cho SY, Ko AR, Kwon MJ, Kim JY, Park HD, Kim OH, Jin DK. Clinical, biochemical, and genetic analysis of two korean patients with trichorhinophalangeal syndrome type I and growth hormone deficiency. Ann Clin Lab Sci. 2012;42(3):307–12.
Stagi S, Bindi G, Galluzzi F, Lapi E, Salti R, Chiarelli F. Partial growth hormone deficiency and changed bone quality and mass in type I trichorhinophalangeal syndrome. Am J Med Genet A. 2008;146A(12):1598-604.
Sidler JA, Filges I, Boesch N, Ramelli GP, Röthlisberger B, Huber AR, Tercanli S, Bronz L, Miny P, Heinimann K. TRPS1 codon 952 constitutes a mutational hot spot in trichorhinophalangeal syndrome type I and could be associated with intellectual disability. Clin Dysmorphol. 2012;21(2):87–90.
Sorting Intolerant From Tolerant. http://sift.jcvi.org. Accessed 26 Jan 2017
Mutation Taster. www.mutationtaster.org. Accessed 26 Jan 2017
Polymorphism Phenotyping v2. http://genetics.bwh.harvard.edu/pph2/. Accessed 26 Jan 2017
Kantaputra P, Miletich I, Lüdecke HJ, Suzuki EY, Praphanphoj V, Shivdasani R, Wuelling M, Vortkamp A, Napierala D, Sharpe PT. Tricho-rhino-phalangeal syndrome with supernumerary teeth. J Dent Res. 2008;87(11):1027–31.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
The Exome Aggregation Consortium. http://exac.broadinstitute.org. Accessed 26 Jan 2017
Exome Variant Server. http://evs.gs.washington.edu/EVS/. Accessed 26 Jan 2017
Single Nucleotide Polymorphism Database. http://www.ncbi.nlm.nih.gov/SNP/. Accessed 26 Jan 2017
Langer Jr LO. The thoracic-pelvic-phalangeal dystrophy. Birth Defects OAS. 1969;5(4):55–64.
Gorlin RJ, Cohen Jr MM, Wolfson J. Tricho-rhino-phalangeal syndrome. Am J Dis Child. 1969;118:595–9.
Nagai T, Nishimura G, Kasai H, Hasegawa T, Kato R, Ohashi H, Fukushima Y. Another family with tricho-rhino-phalangeal syndrome type III (Sugio-Kajii syndrome). Am J Med Genet. 1994;49:278–80.
Vilain C, Sznajer Y, Rypens F, Desir D, Abramowicz MJ. Sporadic case of trichorhinophalangeal syndrome type III in a European patient. Am J Med Genet. 1999;85:495–7.
Parkhurst RD, Light GS, Bacon GE, Gall Jr JC. Tricho-rhino-phalangeal syndrome with hypoglycemia: case report with endocrine function studies. South Med J. 1972;65:457-59.
Walravens PA, Hambridge KM, Wesenberg RI, Greensher A. Endocrine and radiological abnormalities associated with the tricho-rhino-phalangeal (TRP) syndrome [abstract]. Clin Res. 1975; 23147A.
Seitz CS, Lüdecke HJ, Wagner N, Bröcker EB, Hamm H. Trichorhinophalangeal syndrome type I: clinical and molecular characterization of 3 members of a family and 1 sporadic case. Arch Dermatol. 2001;137(11):1437–42.
Shibata A, Tanahashi K, Sugiura K, Akiyama M. TRPS1 Haploinsufficiency Results in Increased STAT3 and SOX9 mRNA Expression in Hair Follicles in Trichorhinophalangeal Syndrome. Acta Derm Venereol. 2015;95(5):620–1.
Suemoto H, Muragaki Y, Nishioka K, Sato M, Ooshima A, Itoh S, et al. Trps1 regulates proliferation and apoptosis of chondrocytes through Stat3 signaling. Dev Biol. 2007;312:572–81.
Gai Z, Gui T, Muragaki Y. The function of TRPS1 in the development and differentiation of bone, kidney, and hair follicles. Histol Histopathol. 2011;26:915–21.
Hilton MJ, Sawyer JM, Gutierrez L, et al. Analysis of novel and recurrent mutationsresponsible for the tricho-rhino-phalangeal syndromes. J Hum Genet. 2002;47:103–6.
Chen LH, Ning CC, Chao SC. Mutations in TRPS1 gene in trichorhinophalangeal syndrome type I in Asian patients. Br J Dermatol. 2010;163:416–9.
Vora NS, Shah S, Dave JN, et al. Trichorhinophalangeal syndrome type I. Indian J Dermatol Venereol Leprol. 1995;61:111–2.
Hussels IE. Trichorhinophalangeal syndrome in two sibs. Birth Defects Orig Artic Ser. 1971;7:301–3.
Corsini C, Gencik M, Willems M, Decker E, Sanchez E, Puechberty J, Schneider A, Girard M, Edery P, Bretonnes P, Cottalorda J, Lefort G, Jeandel C, Sarda P, Genevieve D. Somatic mosaicism in trichorhinophalangeal syndrome: a lesson for genetic counseling. Eur J Hum Genet. 2014;22(1):136–9.
Acknowledgements
The authors would like to thank the family who agreed to publication of their clinical details for the benefit of other families.
Funding
This study had no funding source.
Availability of data and materials
All data is contained in the manuscript.
Authors’ contributions
WS: drafted the manuscript and participated in the collection of clinical data and conceived figures. SCE: Conceived the study and its coordination and helped in drafting the manuscript. SM: carried out the molecular genetic studies. MZ: participated in the design of the study and provided DNA samples. AS: Supervised the study design with clinical data collection. KH: Coordinated the study, supervised the molecular genetic studies and revised the manuscript critically. All authors read and approved the final manuscript.
Competing interest
The authors declare that they have no competing interests.
Consent for publication
Each family member gave written consent for clinical data and images to be published (Parents gave written consent on behalf of patient 1).
Ethics approval and consent to participate
Ethics approval is deemed unnecessary according to national regulations. This case report was not presented as a research study because all family members were seen in a medical consultation for a diagnostic purpose and they gave their written consent to participate and benefit from molecular analysis, the parents gave their written consent on behalf of patient 1.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Smaili, W., Elalaoui, S.C., Meier, S. et al. A novel TRPS1 mutation in a Moroccan family with Tricho-rhino-phalangeal syndrome type III: case report. BMC Med Genet 18, 50 (2017). https://doi.org/10.1186/s12881-017-0413-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0413-8