
Abstract
Background
Sepsis is a life-threatening condition that is characterized by multiorgan dysfunction and caused by dysregulated cytokine networks, which are closely associated with sepsis progression and outcomes. However, currently available treatment strategies that target cytokines have failed. Thus, this study aimed to investigate the interplay between genetically predicted circulating concentrations of cytokines and the outcomes of sepsis and to identify potential targets for sepsis treatment.
Methods
Data related to 35 circulating cytokines in 31,112 individuals (including 11,643 patients with sepsis) were included in genome-wide association studies (GWASs) from the UK Biobank and FinnGen consortia. A bidirectional two-sample Mendelian randomization (MR) analysis was performed using single nucleotide polymorphisms (SNPs) to evaluate the causal effects of circulating cytokines on sepsis outcomes and other cytokines.
Results
A total of 35 inflammatory cytokine genes were identified in the GWASs, and 11 cytokines, including Interleukin-1 receptor antagonist (IL-1ra), macrophage inflammatory protein 1 (MIP1α), IL-16, et al., were associated with sepsis outcome pairs according to the selection criteria of the cis-pQTL instrument. Multiple MR methods verified that genetically predicted high circulating levels of IL-1ra or MIP1α were negatively correlated with genetic susceptibility to risk of sepsis, including sepsis (28-day mortality), septicaemia, streptococcal and pneumonia-derived septicaemia (P ≤ 0.01). Furthermore, genetic susceptibility of sepsis outcomes except sepsis (28-day mortality) markedly associated with the circulating levels of five cytokines, including active plasminogen activator inhibitor (PAI), interleukin 7 (IL-7), tumour necrosis factor alpha (TNF-α), beta nerve growth factor (NGF-β), hepatic growth factor (HGF) (P < 0.05). Finally, we observed that the causal interaction network between MIP1α or IL-1ra and other cytokines (P < 0.05).
Conclusions
This comprehensive MR analysis provides insights into the potential causal mechanisms that link key cytokines, particularly MIP1α, with risk of sepsis, and the findings suggest that targeting MIP1α may be a potential strategy for preventing sepsis.
Similar content being viewed by others


Introduction
Sepsis is considered a life-threatening systemic disease that arises due to a dysregulated inflammatory response to infection and leads to multiorgan dysfunction, which is the main cause of mortality in patients with severe cases [1]. Although substantial progress has been made in the timely use of antibiotics, fluid resuscitation, and organ support therapies in recent years, the mortality rate of patients with sepsis still reaches in 30% in intensive care units (ICUs) in hospitals [2,3,4,5]. Thus, the discovery of new approaches to prevent the onset and progression of sepsis and its complications is important.
The pathogenesis of sepsis has been extensively explored. Under physiological conditions, the proper balance between proinflammatory and anti-inflammatory effects is critical for immune homeostasis [6]. However, after sepsis onset, the upregulation of proinflammatory cytokines (IL-1β, IL-6, IFN-γ and TNF-ɑ) and chemokines (MCP-1/CCL2 and MIP1α/CCL3) that are generated by inflammatory cells is critical for eliminating infectious pathogens and quickly restoring immune balance, whereas excessive inflammation can cause tissue and organ damage [7]. If pathogens are not cleared in a timely manner, they can cause immune cell dysfunction and the release of large amounts of anti-inflammatory cytokines (IL-4, IL-10, and IL-37), leading to persistent immunosuppression, which is the main factor contributing to the poor prognosis and risk of death in septic patients [8, 9]. Thus, immune homeostasis plays a key role in the pathophysiology of sepsis and in determining clinical outcomes. The mechanisms regulating the release of inflammatory cytokines and chemokines in sepsis are complex. Several studies have indicated that genetic variations affect the host immune response to infection [10, 11]. More importantly, each cytokine has the potential to serve as a drug target. Unfortunately, all clinical trials that have aimed to inhibit the inflammatory response or target cytokines have failed [12]; thus, the impact of genetically determined levels of inflammatory factors on sepsis must be determined.
Genome-wide association studies (GWASs) are key approaches for identifying genetic factors that are related to disease severity and survival prognosis in patients with several types of infectious diseases. Several GWASs have identified hundreds of single-nucleotide polymorphisms (SNPs) that are associated with sepsis-related inflammatory cytokines and outcomes [13,14,15]. This information creates the opportunity to investigate potentially causal genetic relationships between these SNPs and other clinically relevant sepsis traits via a Mendelian randomization (MR) approach [16]. Indeed, the first sepsis-related GWAS indicated that three independent low-frequency variants in SAMD9 (a possible mediator of the inflammatory response) are associated with reduced 28-day survival in septic patients [17]. Moreover, the serum levels of multiple inflammatory cytokines, such as IL-6 and TNF-α, are positively associated with the risk and outcomes of sepsis [18, 19]. However, whether these associations are causal and whether circulating cytokines could serve as potential therapeutic targets for sepsis are unclear.
In the present study, via bidirectional MR analysis, we examined the potentially causal links between circulating cytokines and sepsis outcomes to identify potential therapeutic targets. Moreover, we studied the effects of sepsis traits on the levels of these circulating cytokines. Additionally, we constructed a protein‒protein interaction (PPI) network of the causal genes and defined the effects of circulating cytokines on the levels of other cytokines.
Methods
Study overview
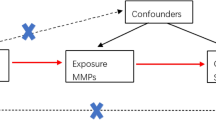
A bidirectional MR analysis approach was used in this study to examine the causative effects of cytokines on sepsis and its outcomes (Fig. 1). First, we assessed the causal associations between the levels of 35 cytokines predicted by genetics and the risk of sepsis. We subsequently conducted reverse MR analysis to determine whether the genetic prediction of sepsis risk influences the levels of 35 cytokines. We derived causal estimates for the associations of serum cytokines with the risk of sepsis via four different MR approaches on the basis of multiple serum cytokine-associated genetic variants. We used summary estimates for the effects of genotype on exposures and outcomes from the largest available meta-analyses of previous GWASs, focusing on datasets including participants predominantly of European descent, to ensure consistent allele frequencies across the datasets and avoid possible modifications of genetic effects by ancestral origin (Table 1). The present study was conducted following the STROBE-MR guidelines for transparent reporting of MR studies.

Schematic representation of the study. The analyses conducted in this study are numbered. Genetic variants are predetermined at birth, are largely randomly assorted in a population, and can be used as instrumental variables to estimate the causal association of an exposure (e.g., cytokines) with an outcome of interest (e.g., sepsis). First, the genetic prediction of cytokine levels was used as the exposure, and sepsis was the outcome to assess whether changes in cytokine levels affect sepsis outcomes. The genetic prediction of risk of sepsis was then used as the exposure, and cytokine levels were used as the outcome to evaluate whether sepsis affects cytokine levels. This approach relies on three assumptions: the genetic variants must be associated with the exposure, the genetic variants must not be associated with confounders, and the genetic variants must influence the risk of the outcome through the exposure and not through other pathways. GWAS = genome-wide association study
Datasets
The current analysis involved the use of publicly available deidentified data. All the original studies included appropriate ethical reviews and provided informed consent. The GWAS summary data for cytokines were sourced from Bouras et al. (2022) [16] and Karhunen et al. (2023) [20]. The analytic sample of sepsis outcomes included up to 486,484 individuals of European ancestry from the UK Biobank and 208,794 individuals of European ancestry from the FinnGen consortium (Table 1). A total of 6 sepsis traits were included in the analysis, with 362 to 11,643 individuals in the patient group and 197,660 to 484,588 individuals in the control group. The trait name is consistent with the original data used. In addition, the population studied for cytokines does not overlap with the population studied for sepsis.
Development of a genetic instrument
This MR study was designed to use summary statistics from large-scale GWASs (Table 1). A total of SNPs that were associated with serum cytokine levels were identified in a meta-analysis of individuals of European origin [20]. The Mendelian randomization analyses that used circulating cytokine concentrations as exposures were performed with 31 cis-pQTL and 27 cis-eQTL cytokine (a total of 35 unique cytokines) instruments. These SNPs were associated with serum cytokine levels at the genome-wide significance level (P < 1.0 × 10− 4 for pQTLs and P < 0.05 for eQTLs). Variants with a minor allele frequency (MAF) < 0.05 were excluded. In the context of a cis-region MR, using a very small correlation threshold may result in a loss of causal variants; therefore, clumping was performed using a pairwise linkage disequilibrium (LD) threshold of r2 < 0.1 [21]. The circulating cytokine levels were predicted via a set of estimated SNP effects based on a GWAS. A total of 375 independent SNPs were ultimately selected as proxies for the circulating levels of 35 cytokines. Detailed information on the SNPs that were used is presented in Supplementary Table S1. Instruments for each of the six sepsis traits were selected as SNPs that were associated with that trait at P < 5 × 10− 6 and were uncorrelated (r2 < 0.001). When the reverse MR analysis was conducted, only 35 cytokines were included to correspond to the forwards MR analysis.
Gene interactions and epigenetic effects
Gene interactions and networks were analysed via the GeneMANIA prediction server (v3.5.1) (http://genemania.org, accessed on 28 April 2024) [22] and plotted via Cytoscape (v3.6.1). Pathway-based association testing was achieved by defining a biological pathway incorporating the gene targets of interest.
MR analyses
The primary analysis was conducted via a random-effects inverse-variance-weighted method [23]. The secondary MR methods include MR‒Egger [24], simple median, and weighted median [25] methods. The weighted median method can reduce the bias of valid inverse variances and is suitable for applications involving multiple genetic analyses. MR‒Egger regression can detect and adjust for pleiotropy, but the precision of the estimates is low. Z scores (the beta value of continuous traits or log odds value of the disease risk divided by the standard error of the cross-trait associations) obtained from MR analyses were used to construct a heatmap.
Sensitivity analyses
We conducted multiple sensitivity analyses with respect to the MR tests to exclude possible biases (horizontal pleiotropy, i.e., variants included in the genetic instrument having an effect on the disease outside their effects on the exposure in MR) under different scenarios in the MR estimates and to increase the reliability of the MR results [26]. Finally, a modified Cochran Q statistic and leave-one-out analysis were conducted to detect heterogeneity in the results [26]. As a result of these different methods used to compare the results, better agreement and higher reliability were obtained.
Statistical analyses
MR analyses were conducted with the TwoSampleMR R package [23]. All the statistical tests were two-sided. All the statistical analyses were conducted with R version 4.3 (R Foundation).
Results
Circulating inflammatory cytokines in GWASs
A total of 35 inflammatory cytokine genes were identified in the GWAS, and these cytokines are shown in Tables S1. The genetic association estimates for these SNPs were used for the following MR analyses.
Effects of circulating cytokines on sepsis traits
We analysed all SNPs in inflammatory cytokine-related genes and 6 sepsis-related outcomes from the UK Biobank and FinnGen consortia to determine whether circulating cytokines affect sepsis traits, and the results revealed that a total of 312 SNPs were associated with the serum levels of 35 cytokines after screening (Table S2). We then used the IVW method for the MR analysis. According to the selection criteria of the cis-pQTL instrument, the associations of eleven cytokines (including IL-1ra, IL12p70, IL-16, IL-18, GROa, MIP1α, MCSF, sICAM, VEGF, SCF, and TRAIL) with sepsis outcome pairs were significant (P < 0.05, Fig. 2). Genetically predicted high circulating levels of IL-1 receptor antagonist (IL-1ra) and macrophage inflammatory protein 1 alpha (MIP1α) were negatively correlated with genetic susceptibility to sepsis risk, including septicaemia, and pneumonia-derived septicaemia (P < 0.002), after Bonferroni correction. (Fig. 2). Furthermore, we performed MR analysis via cis-eQTL instruments. The associations of 4 cytokines, namely, IL-1ra, beta-nerve growth factor (bNGF), stem cell factor (SCF) and macrophage colony-stimulating factor (MCSF), with sepsis outcome pairs were nominally significant (P < 0.05) (Figure S1).

Mendelian randomization effect size estimates (z scores) of genetically predicted circulating cytokine levels on sepsis traits on the basis of the definition of the cis-pQTL instrument. Square tiles indicate that the association is nominally significant (P < 0.05). The asterisks indicate significance after the Bonferroni correction for testing multiple cytokines (P < 0.002 [0.05/number of cytokines])
Associations of inflammatory cytokines with sepsis traits
We used multiple MR methods to further explore the associations of IL-1ra and MIP1α with various sepsis traits, and the associations were significant according to the MR results (P < 0.05; Fig. 3). Genetically predicted high circulating levels of IL-1ra were negatively correlated with genetic susceptibility to risk of sepsis, including septicaemia [OR = 0.85 (95% CI: 0.77–0.94, P = 1.91 × 10− 3)] and streptococcal septicaemia [OR = 0.73 (95% CI: 0.57–0.93, P = 0.012)]. Furthermore, genetically predicted high circulating levels of MIP1α were also negatively correlated with genetic susceptibility to risk of sepsis, including sepsis [OR = 0.93 (95% CI: 0.89–0.98, P = 3.08 × 10− 3)], sepsis (28-day mortality) [OR = 0.85 (95% CI: 0.76–0.95, P = 4.63 × 10− 3)], septicaemia [OR = 0.91 (95% CI: 0.84–0.99, P = 0.022)], and pneumonia-derived septicaemia [OR = 0.65 (95% CI: 0.51–0.83, P = 4.77 × 10− 4)]. These findings suggest that these two cytokines play a role in protecting against the risk of sepsis. All the results of this analysis are shown in Table S3. We also conducted heterogeneity and pleiotropy tests to ensure the reliability of the associations between MIP1α levels and sepsis traits. Cochran Q tests for IVW (P = 0.86 for sepsis, P = 0.95 for sepsis (28-day death) indicated no heterogeneity in the SNPs that were included in the study. No outliers were identified in the leave-one-out analysis (Figure S2 and S3).

Mendelian randomization analysis of MIP1α and IL-1ra levels associated with the risks of sepsis traits. Significant differences in the associations are shown (P < 0.05). All the results of this analysis are shown in Table S3
Effects of sepsis traits on circulating inflammatory cytokine levels
MR IVW analysis revealed no significant associations (P < 0.0012) between genetic susceptibility to sepsis and circulating levels of cytokines (Fig. 4). Moreover, a negative association was observed between genetic susceptibility to sepsis and circulating levels of active plasminogen activator inhibitor (PAI) [OR = 0.811 (95% CI: 0.682–0.965, P = 0.018)]. A positive association was observed between streptococcal septicaemia and circulating levels of interleukin 7 (IL-7) [OR = 1.056 (95% CI: 1.007–1.108, P = 0.025)] or tumour necrosis factor alpha (TNFα) [OR = 1.321 (95% CI: 1.008–1.732, P = 0.044)] and between pneumonia-derived septicaemia and circulating levels of beta nerve growth factor (beta NGF) [OR = 1.077 (95% CI: 1.009–1.149, P = 0.025)]. In addition, genetic susceptibility to streptococcal septicaemia was related to circulating levels of hepatic growth factor (HGF) [OR = 0.941 (95% CI: 0.891–0.993, P = 0.027)] (Fig. 4; Table S4). Interestingly, these cytokines do not overlap with the cytokines that were previously shown to lead to sepsis and its adverse outcomes.

Mendelian randomization effect size estimates (z scores) of genetically predicted sepsis traits on circulating cytokine levels. Square tiles indicate that the association is nominally significant (P < 0.05). All the results of this analysis are shown in Table S4
Causal linkage network among circulating levels of MIP1α and IL-1ra and other circulating cytokines
Evidence from the Mendelian randomization analysis (P < 0.05) revealed associations between MIP1α, IL-1ra and other cytokines when the cis-pQTL instrument was used, with |β| values ranging from 0.05 to 0.60 (Fig. 5). The results revealed a dense interaction network between MIP1α and IL-1ra and other cytokines. The strongest associations were noted for genetically predicted MIP1α levels with circulating levels of thirteen (|β| from 0.05 to 0.60) other cytokines and for IL-1ra levels with the levels of six (|β| from 0.07 to 0.44) other cytokines.

Mendelian randomization results for the effects of genetically predicted MIP1α and IL-1ra concentrations on the circulating levels of other cytokines when considering cis-protein instruments. The results are plotted only for effects with P < 0.05. Association estimates are indicated with lines: red represents positive correlations, and blue represents negative correlations. The arrow starts with the exposure factor and points to the outcome factor
Functional analysis of the IL-1ra and MIP1α genes
Finally, we tested whether genetically proxied cytokines (IL1ra, encoded by IL1RN, and MIP1α/CCL3) influence other inflammatory cytokine-related genes. Gene coexpression network analysis revealed that IL1RN and MIP1α had high network connectivity with a subset of sepsis-related genes, including IL-1R2, CCR4, ACKR2, CXCL8, and IL1R1 (Figure S4). The functional enrichment analysis suggested that these genes were involved in physical interactions, coexpression, prediction, genetic interactions, pathways, shared protein domains, and colocalization and were enriched mainly in response to IL-1, the cellular response to molecules of bacterial origin, the response to lipopolysaccharides, cytokine receptor activity, and the cellular response to biotic stimuli (Figure S4).
Discussion
In the present study, via GWAS data and MR analysis, we provide comprehensive genetic evidence for the effects of genetic proxies of circulating inflammatory cytokines on sepsis outcomes. Our results revealed a negative association of genetically predicted circulating MIP1α and IL-1ra levels with pneumonia-derived septicaemia in patients with sepsis. We also identified new therapeutic targets (IL-1ra and MIP1α) for sepsis and related complications. Our data also indicate that sepsis outcomes affect the circulating levels of cytokines and that a dense interaction network exists between cytokines.
Circulating inflammatory cytokines are influential mediators of immune and inflammatory reactions. High circulating levels of cytokines are involved in sepsis onset and are major causes of high mortality in patients with severe sepsis [18]. Although blocking the activities of proinflammatory cytokines can significantly increase survival in animal models of sepsis, a similar therapeutic strategy has not improved patient outcomes [27], and drugs that target TNF-α, IL-1β, or Toll-like receptors have not achieved satisfactory clinical results in improving the survival rate of patients with sepsis [28, 29]. This finding was consistent with the findings of the present study, which indicate that, except MIP1ɑ, most cytokines are not strongly correlated with sepsis or sepsis prognosis (Fig. 2), suggesting that no causal link exists between most cytokines and sepsis; however, these results do not exclude the role of the cytokine storm in the pathogenesis of sepsis. Additionally, at the cis-eQTL level, MR results revealed a nominally significant association between IL1ra levels and sepsis outcomes, whereas MIP1α levels did not show a significant association, possibly because only one SNP was used as the instrumental variable after screening. Cytokine storms also occur in several pathological conditions, such as viral infections, cancer, and multiorgan failure [30], and might be manifestations of an aberrant internal environment during sepsis rather than causal factors [31]. This hypothesis must be validated in future research.
MIP1α (also known as CCL3) is a member of the C-C chemokine family that is secreted by monocytes, macrophages, and T cells. The primary functions of MIP1α are the activation and chemotaxis of leukocytes, B cells, CD8+ T cells and eosinophils under inflammatory conditions [32]. In addition, MIP1α can induce the secretion of multiple inflammatory cytokines (TNFα, IL-6 and IL-1) [33], [34, 35]. Macrophages, but not neutrophils, are the major effector cells, and MIP1α is needed for host resistance against bacterium-induced sepsis and other immune diseases [36]. Compared with peripheral blood mononuclear cells (PBMCs) from healthy donors, PBMCs that are isolated from severely burned patients are unable to release MIP1α in culture after stimulation with an anti-human CD3 monoclonal antibody (mAb). These data indicate that MIP1α plays a beneficial role in infectious complications in burn patients [37]. Moreover, MIP1ɑ and MIP-1β are upregulated during acute inflammation, and MIP1ɑ can mediate fever in animals independent of cyclooxygenase blockade by ibuprofen. Interestingly, MIP1 levels were not influenced by blocking circulating TNF-α in a human study [38]. Several studies have indicated that MIP1α has protective effects on sepsis and death [36]. In addition, increased expression levels of MIP-1α are negatively associated with the progression of multiple inflammatory diseases, such as multiple myeloma, lung cancer, multiple sclerosis, HIV infection, and allergic asthma [39, 40]. Therefore, MIP1α has become a cytokine of interest for studying the pathophysiology of sepsis and its complications. Our results revealed a causal association between circulating IL-1ra and MIP1α levels and sepsis traits (Fig. 2) and suggested that the upregulation of IL-1ra and MIP1ɑ may have beneficial effects on improving sepsis outcomes.
We explored whether sepsis traits affect circulating cytokine levels after the onset of sepsis to investigate the impact of reverse causality. Interestingly, we found a correlation between genetically predicted sepsis traits and the circulating levels of cytokines (including IL-7, HGF, beta NGF, TNFα, and active PAI). This finding is consistent with the results of many observational studies [41,42,43]. These results suggest a bidirectional causal relationship between cytokines and sepsis and its outcomes. Although these reverse-causal-associated cytokines do not overlap with the associated proinflammatory cytokines, this result does not negate the value of other cytokines in the progression of sepsis, as a dense causal network exists between the two key cytokines and the levels of other cytokines (Fig. 5). Further research is needed to elucidate the specific mechanisms underlying the complex network of these cytokines and the progression of sepsis [44].
This study has several limitations. The predominantly European ancestry of the subjects limits the generalizability of the results to other ethnic groups. As with all MR studies, we could not address unobserved pleiotropy. Furthermore, a potential nonlinear association between serum cytokine levels and sepsis could not be evaluated. In addition, we only considered changes in cytokine levels as a result of genetic factors, which accounted for only a small proportion of the changes in cytokine levels. The regulatory network formed by these cytokines is a complex mechanism that has not been fully explored here.
In conclusion, we used MR analyses and large-scale genetic data to explore the associations of 35 circulating inflammatory cytokines with sepsis and its outcomes. Our data revealed several significant associations between circulating cytokines, particularly MIP1α, and sepsis outcomes, and it identified potential therapeutic targets, including circulating IL-1ra and MIP1ɑ, for reducing sepsis risk. Future studies are needed to confirm the genetic effects on sepsis survival, and future studies should validate the findings in multiple studies with larger sample sizes to evaluate the roles of cytokines in regulating macrophage chemotaxis during sepsis and to validate the potential of these cytokines as targets for sepsis prevention.
Data availability
The datasets supporting the conclusions of this article are included within the article and its additional files.
References
Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet (London England). 2018;392(10141):75–87.
Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, Angus DC, Reinhart K. International Forum of Acute Care T: Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current estimates and limitations. Am J Respir Crit Care Med. 2016;193(3):259–72.
Stevenson EK, Rubenstein AR, Radin GT, Wiener RS, Walkey AJ. Two decades of mortality trends among patients with severe sepsis: a comparative meta-analysis*. Crit Care Med. 2014;42(3):625–31.
Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41(5):1167–74.
Liu YC, Yao Y, Yu MM, Gao YL, Qi AL, Jiang TY, Chen ZS, Shou ST, Chai YF. Frequency and mortality of sepsis and septic shock in China: a systematic review and meta-analysis. BMC Infect Dis. 2022;22(1):564.
van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 2017;17(7):407–20.
Chaudhry H, Zhou J, Zhong Y, Ali MM, McGuire F, Nagarkatti PS, Nagarkatti M. Role of cytokines as a double-edged sword in sepsis. In vivo (Athens Greece). 2013;27(6):669–84.
Barichello T, Generoso JS, Singer M, Dal-Pizzol F. Biomarkers for sepsis: more than just fever and leukocytosis-a narrative review. Crit Care. 2022;26(1):14.
Tan K, Minejima E, Lou M, Mack WJ, Nieberg P, Wong-Beringer A. Cytokine measurements add value to clinical variables in predicting outcomes for Staphylococcus aureus bacteremia. BMC Infect Dis. 2021;21(1):317.
Andreakos E, Abel L, Vinh DC, Kaja E, Drolet BA, Zhang Q, O’Farrelly C, Novelli G, Rodriguez-Gallego C, Haerynck F, et al. A global effort to dissect the human genetic basis of resistance to SARS-CoV-2 infection. Nat Immunol. 2022;23(2):159–64.
Netea MG, Wijmenga C, O’Neill LA. Genetic variation in toll-like receptors and disease susceptibility. Nat Immunol. 2012;13(6):535–42.
Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. 2017;39(5):517–28.
Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran P, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558(7708):73–9.
Folkersen L, Gustafsson S, Wang Q, Hansen DH, Hedman AK, Schork A, Page K, Zhernakova DV, Wu Y, Peters J, et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat Metab. 2020;2(10):1135–48.
Guillen-Guio B, Lorenzo-Salazar JM, Ma SF, Hou PC, Hernandez-Beeftink T, Corrales A, Garcia-Laorden MI, Jou J, Espinosa E, Muriel A, et al. Sepsis-associated acute respiratory distress syndrome in individuals of European ancestry: a genome-wide association study. Lancet Respir Med. 2020;8(3):258–66.
Bouras E, Karhunen V, Gill D, Huang J, Haycock PC, Gunter MJ, Johansson M, Brennan P, Key T, Lewis SJ, et al. Circulating inflammatory cytokines and risk of five cancers: a mendelian randomization analysis. BMC Med. 2022;20(1):3.
Hernandez-Beeftink T, Guillen-Guio B, Lorenzo-Salazar JM, Corrales A, Suarez-Pajes E, Feng R, Rubio-Rodriguez LA, Paynton ML, Cruz R, Garcia-Laorden MI, et al. A genome-wide association study of survival in patients with sepsis. Crit Care. 2022;26(1):341.
Li XY, Liu M, Fu YJ, Jiang YJ, Zhang ZN. Alterations in levels of cytokine following treatment to predict outcome of sepsis: a meta-analysis. Cytokine. 2023;161:156056.
Georgescu AM, Banescu C, Azamfirei R, Hutanu A, Moldovan V, Badea I, Voidazan S, Dobreanu M, Chirtes IR, Azamfirei L. Evaluation of TNF-alpha genetic polymorphisms as predictors for sepsis susceptibility and progression. BMC Infect Dis. 2020;20(1):221.
Karhunen V, Gill D, Huang J, Bouras E, Malik R, Ponsford MJ, Ahola-Olli A, Papadopoulou A, Palaniswamy S, Sebert S, et al. The interplay between inflammatory cytokines and cardiometabolic disease: bi-directional mendelian randomisation study. BMJ Med. 2023;2(1):e000157.
Gkatzionis A, Burgess S, Newcombe PJ. Statistical methods for cis-mendelian randomization with two-sample summary-level data. Genet Epidemiol. 2023;47(1):3–25.
Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(Suppl 1):S4.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018, 7.
Burgess S, Thompson SG. Interpreting findings from mendelian randomization using the MR-Egger method. Eur J Epidemiol 2017, 32(5):377–89.
Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from mendelian randomization analyses with multiple genetic variants. Epidemiology. 2017;28(1):30–42.
Hemani G, Bowden J, Davey Smith G. Evaluating the potential role of pleiotropy in mendelian randomization studies. Hum Mol Genet. 2018;27(R2):R195–208.
Brown KA, Brown GA, Lewis SM, Beale R, Treacher DF. Targeting cytokines as a treatment for patients with sepsis: a lost cause or a strategy still worthy of pursuit? Int Immunopharmacol. 2016;36:291–9.
Cohen J, Opal S, Calandra T. Sepsis studies need new direction. Lancet Infect Dis. 2012;12(7):503–5.
Angus DC. The search for effective therapy for sepsis: back to the drawing board? JAMA. 2011;306(23):2614–5.
Karki R, Kanneganti TD. The ‘cytokine storm’: molecular mechanisms and therapeutic prospects. Trends Immunol. 2021;42(8):681–705.
Bosmann M, Ward PA. The inflammatory response in sepsis. Trends Immunol. 2013;34(3):129–36.
Cook DN, Smithies O, Strieter RM, Frelinger JA, Serody JS. CD8 + T cells are a biologically relevant source of macrophage inflammatory protein-1 alpha in vivo. J Immunol. 1999;162(9):5423–8.
Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392(6676):565–8.
Cook DN. The role of MIP-1 alpha in inflammation and hematopoiesis. J Leukoc Biol. 1996;59(1):61–6.
Standiford TJ, Kunkel SL, Lukacs NW, Greenberger MJ, Danforth JM, Kunkel RG, Strieter RM. Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak, and early mortality in murine endotoxemia. J Immunol. 1995;155(3):1515–24.
Takahashi H, Tashiro T, Miyazaki M, Kobayashi M, Pollard RB, Suzuki F. An essential role of macrophage inflammatory protein 1alpha/CCL3 on the expression of host’s innate immunities against infectious complications. J Leukoc Biol. 2002;72(6):1190–7.
Kobayashi M, Takahashi H, Sanford AP, Herndon DN, Pollard RB, Suzuki F. An increase in the susceptibility of burned patients to infectious complications due to impaired production of macrophage inflammatory protein 1 alpha. J Immunol. 2002;169(8):4460–6.
O’Grady NP, Tropea M, Preas HL 2nd, Reda D, Vandivier RW, Banks SM, Suffredini AF. Detection of macrophage inflammatory protein (MIP)-1alpha and MIP-1beta during experimental endotoxemia and human sepsis. J Infect Dis. 1999;179(1):136–41.
Guazzone VA, Jacobo P, Theas MS, Lustig L. Cytokines and chemokines in testicular inflammation: a brief review. Microsc Res Tech. 2009;72(8):620–8.
Ntanasis-Stathopoulos I, Fotiou D, Terpos E. CCL3 signaling in the Tumor Microenvironment. Adv Exp Med Biol. 2020;1231:13–21.
Gharamti AA, Samara O, Monzon A, Montalbano G, Scherger S, DeSanto K, Chastain DB, Sillau S, Montoya JG, Franco-Paredes C, et al. Proinflammatory cytokines levels in sepsis and healthy volunteers, and tumor necrosis factor-alpha associated sepsis mortality: a systematic review and meta-analysis. Cytokine. 2022;158:156006.
Peng F, Liang C, Chang W, Sun Q, Xie J, Qiu H, Yang Y. Prognostic significance of plasma hepatocyte growth factor in Sepsis. J Intensive Care Med. 2022;37(3):352–8.
Hillenbrand A, Knippschild U, Weiss M, Schrezenmeier H, Henne-Bruns D, Huber-Lang M, Wolf AM. Sepsis induced changes of adipokines and cytokines - septic patients compared to morbidly obese patients. BMC Surg. 2010;10:26.
Jansen JE, Aschenbrenner D, Uhlig HH, Coles MC, Gaffney EA. A method for the inference of cytokine interaction networks. PLoS Comput Biol. 2022;18(6):e1010112.
Acknowledgements
None.
Funding
This work was supported by grants from the Open Project of Beijing Key Laboratory of Cardiopulmonary Cerebral Resuscitation, Beijing Chao-Yang Hospital.
Author information
Authors and Affiliations
Contributions
Conceptualization, methodology, investigation, data curation, validation, writing—original draft preparation, W.J.; Supervision, project administration, writing—review and editing, and funding acquisition, H.L. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
Ethical approval and informed consent were not required for the present study, as they were obtained in the original studies.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Disclosures
None of the funding organizations were involved in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication. The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, WX., Li, HH. Circulating inflammatory cytokines and the risk of sepsis: a bidirectional mendelian randomization analysis. BMC Infect Dis 24, 793 (2024). https://doi.org/10.1186/s12879-024-09689-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-024-09689-z