
Abstract
Background
Since the emergence of SARS-CoV-2, evolutionary pressure has driven large increases in the transmissibility of the virus. However, with increasing levels of immunity through vaccination and natural infection the evolutionary pressure will switch towards immune escape. Genomic surveillance in regions of high immunity is crucial in detecting emerging variants that can more successfully navigate the immune landscape.
Methods
We present phylogenetic relationships and lineage dynamics within England (a country with high levels of immunity), as inferred from a random community sample of individuals who provided a self-administered throat and nose swab for rt-PCR testing as part of the REal-time Assessment of Community Transmission-1 (REACT-1) study. During round 14 (9 September–27 September 2021) and 15 (19 October–5 November 2021) lineages were determined for 1322 positive individuals, with 27.1% of those which reported their symptom status reporting no symptoms in the previous month.
Results
We identified 44 unique lineages, all of which were Delta or Delta sub-lineages, and found a reduction in their mutation rate over the study period. The proportion of the Delta sub-lineage AY.4.2 was increasing, with a reproduction number 15% (95% CI 8–23%) greater than the most prevalent lineage, AY.4. Further, AY.4.2 was less associated with the most predictive COVID-19 symptoms (p = 0.029) and had a reduced mutation rate (p = 0.050). Both AY.4.2 and AY.4 were found to be geographically clustered in September but this was no longer the case by late October/early November, with only the lineage AY.6 exhibiting clustering towards the South of England.
Conclusions
As SARS-CoV-2 moves towards endemicity and new variants emerge, genomic data obtained from random community samples can augment routine surveillance data without the potential biases introduced due to higher sampling rates of symptomatic individuals.
Similar content being viewed by others


Background
Since its first documented case in India in November 2020 [1] the Delta variant of SARS-CoV-2 has spread rapidly across the world and by 16 November 2021 was responsible for 99.7% of all SARS-CoV-2 infections [2]. Its rapid rise to dominance has been attributed to greater levels of transmissibility [3, 4] than previously circulating variants with the reproduction number estimated to be over two-fold higher [5], as well as possible reduced vaccine effectiveness against infection [6]. Since its global dissemination, continued adaptive evolution has led to a diverse set of Delta sub-lineages, with distinct combinations of mutations (especially on the spike protein) [7, 8].
Since July 2021 the lineage AY.4.2 (Pango nomenclature [9]), a descendant of the original Delta variant (henceforth B.1.617.2) has increased in proportion in routine surveillance data for England from 8.5% the week beginning 4 October [10] to 14.7% the week beginning 31 October [11]. AY.4.2 was declared a variant under investigation (VUI) by the UK Health Security Agency on 20 October 2021 [12]. Globally AY.4.2 had been detected in 43 countries by 22 November 2021 [13] but had only been estimated at a cumulative proportion greater than 1% in Poland [14]. AY.4.2 has two defining mutations in the spike protein, Y145H and A222V, but is otherwise similar to AY.4, a lineage that is far more widespread. AY.4 was the most prevalent lineage in England on 29 October 2021 [11] and had been detected in 87 countries by 22 November 2021 [15], in some of which it had already been reported as the most prevalent lineage (by 23 November 2021) [16, 17].
England has recorded high levels of SARS-CoV-2 infection over the course of the pandemic [4, 18] and vaccinated, as part of its mass vaccination campaign (Pfizer/BioNTec, Oxford/AstraZeneca and Moderna), a large proportion of its population (80.3% of over 12 year olds double vaccinated by 27 November 2021), with further booster jabs (Pfizer/BioNTec or Moderna) being rolled out in adults (30.5% of over 12 year olds having received a booster dose by 27 November 2021) [18]. This has led to high levels of antibodies against coronavirus with 92.8% of adults in England estimated to test positive for antibodies (IgG antibodies against the SARS-CoV-2 trimeric spike protein) in the week beginning 1 November 2021 [19]. With high vaccination coverage in the population it is likely that there is substantial selective pressure on SARS-CoV-2 towards immune escape and vaccine breakthrough infections. Genomic surveillance in highly immunised regions is crucial to detect emerging variants that can more successfully navigate the immune landscape that has been created by both natural infection and vaccination.
The REal-time Assessment of Community Transmission-1 (REACT-1) study is a series of cross-sectional surveys of the population of England that seeks to estimate the prevalence of SARS-CoV-2 on a monthly basis [4, 20], with genomic sequencing performed on all positive samples with a low enough cycle threshold (Ct) value (a proxy for viral load) and high enough volume. Due to its sampling procedure it does not suffer from the biases of routine surveillance that can be heavily biased towards symptomatic individuals [21]; symptom status can be highly dependent on levels of immunity [22]. Here we present the genomic analysis of the (N = 2163) positive samples for round 14 and round 15 which were collected from 9 to 27 September 2021 and 19 October to 5 November 2021 respectively.
Material and methods
Viral genome sequencing
The methods of the REACT-1 study have been described elsewhere [23]. REACT-1 is a repeat cross-sectional study whereby in each round a random subset of the English population (selected from the National Health Service general practitioners' patient list) is invited to obtain a self-administered swab test (parent/guardian administered for 5–12 year olds). These tests are then sent to a laboratory to undergo rt-PCR testing for the presence of SARS-CoV-2. A round of the study covers a ~ 2- to 3-week period and has occurred approximately monthly since May 2020 with between 100,000 and 185,000 individuals taking part in each round. Since round 8 in January 2021 all positive samples with a low enough N-gene Ct value (the threshold was 34 in rounds 14 and 15 presented here) and sufficient volume have been sent for genome sequencing. Amplification of the extracted RNA was performed using the ARTIC protocol [24] (version 4 primers), with sequence libraries prepared using CoronaHiT [25]; sequencing was performed on the Illumina NextSeq 500 platform. Raw sequences were analysed using the bioinformatic pipeline [26] and then uploaded to CLIMB [27]. Lineages were assigned using PangoLEARN [28] (database version 2021-11-04), a machine learning-based assignment algorithm, using Pango nomenclature [9]. For some sequences of low overall quality, a lineage designation was not possible and so they were not included in the analyses. Samples with less than 50% of bases covered were further excluded from the analysis. Of the 1322 lineages determined during rounds 14 and 15, 1160 individuals provided information on their symptoms in the previous month with 314 (27.1%) reporting no symptoms.
Phylogeographic model
For all sequences from REACT-1 rounds 11 (15 April–3 May 2021), 12 (20 May–7 June 2021), 13 (24 June–12 July 2021), 14 (9 September–27 September 2021) and 15 (19 October–5 November 2021), in which the lineage designated was Delta or a Delta sub-lineage, a maximum likelihood phylogenetic tree was constructed using a HKY model implemented in IQ-TREE [29]. An uncorrelated relaxed clock model implemented in TreeTime [30], assuming a normal distribution of rates with mean 0.0008 substitutions per site per year and a single coalescent rate for the time scale, was then fit to the maximum likelihood phylogenetic tree producing a time-resolved phylogenetic tree. The mutation rates at the tree’s tips were extracted from the model and a Gaussian regression model was fit to the samples obtained during round 14 and 15 for the 8 most prevalent lineages (AY.39, AY.4, AY.4.2, AY.43, AY.44, AY.5, AY.6, B.1.617.2) including lineage and round as covariates. A mugration model (implemented in TreeTime [30]) was run on the time-resolved phylogenetic tree, treating the region in which each sample was isolated as a discrete state. This allowed estimates of the migration rates between regions to be calculated (assumed to be symmetric).
Statistical analyses
The 95% confidence intervals for lineage proportions were calculated using the Wilson method [31] assuming a Binomial distribution. This method is preferred when the number of positives is low but is still valid when this is not the case [32]. Higher accuracy in confidence interval estimates for when the number of positives is low was chosen so that lower bounds on case numbers for rarer lineages were as accurate as possible.
Estimates of the true number of swab-positive infections in England during round 14 and round 15 for lineages in which only one sample was detected in a round were calculated by multiplying the estimated proportion of the lineage for each round, the weighted prevalence estimated for each round [33], and the population size of England [34]. The 95% confidence intervals were estimated by simulating the entire distribution for proportion and weighted prevalence and multiplying the two together. The distribution of weighted prevalence was estimated by randomly sampling from a normal distribution with mean value the central estimate, and standard deviation the width of the 95% confidence interval divided by 3.92 (2 times 1.96). The distribution of the lineage proportion was estimated by calculating the Wilson confidence intervals at different levels (0.00001 to 0.99999 in intervals of 0.00001).
The significance of differences in proportions of particular lineages by age group and region was calculated using Fisher’s exact test with a binary outcome variable (lineage of interest or not). Differences with a p-value less than 0.05 were considered statistically significant. Analysis was only completed for a lineage in a round if there were more than 90 samples (AY.4 round 14, AY.4 round 15, AY.4.2 round 15, B.1.617.2 round 15), so that there were, on average, more than 10 samples per parameter (9 regions in England).
Shannon diversity was calculated using all data for round 14 and round 15, and for each region for round 14 and round 15 [35]. The significance of any differences in Shannon diversity between round 14 and 15 (for all data) and between regions in each round was assessed using the Hutcheson T-test [36] and its associated p-value.
The relative growth rate of a lineage compared to all other lineages was estimated using a Bayesian logistic regression model fit to the binary outcome variable (lineage of interest or not) over time. The two model parameters (intercept and gradient) were given uninformative constant prior distributions. The probability that the growth rate was greater than zero was calculated from the model's posterior. Lineages were deemed to be different to zero if the posterior probability that the growth rate was greater than zero was greater than 0.975 or less than 0.025, similar to a p-value threshold of 0.05.
The growth rates of AY.4.2 and AY.4 infected individuals were estimated by fitting an exponential model to the daily weighted prevalence using all REACT-1 data (all negatives and all AY.4/AY.4.2 associated positives) for rounds 14 and 15 assuming a Binomial likelihood. Weightings for individual REACT-1 samples were calculated using rim weighting [37] by: sex, deciles of the IMD, LTLA counts and ethnic group. Growth rates were then converted to estimates of the reproduction number R assuming a gamma-distributed generation time with the shape parameter, n = 2.29, and rate parameter, b = 0.36 [38] through the equation \((1 + \frac{r}{b}{)}^{n}\) [39]. The multiplicative R advantage of AY.4.2 over AY.4 was estimated using the entire posterior distribution of \({R}_{AY.4.2}/{R}_{AY.4}\) with the median and 95% credible interval reported.
For each lineage with more than 1 sample in a round the presence of clustering was assessed. The pairwise distance matrix between all n samples that were designated to a specific lineage was calculated and from this a mean pairwise distance was calculated for the lineage. Next, 10,000 random combinations of n positive individuals (n positive individuals chosen each time without replacement), for which any lineage was determined, were selected and for each combination the distance matrix and mean distance was calculated. The proportion of the 10,000 estimated mean distances below the lineage-specific mean distance was then calculated. Clustering was deemed to be significant if this proportion was less than 0.05.
For the 8 most prevalent lineages across rounds 14 and 15 Gaussian regression was performed to estimate the mean N- and E-gene Ct values for each lineage and p-values used to assess the significance of any difference to the reference lineage (AY.4). Models were run on all data (rounds 14 and 15 combined) and then run on data from each individual round as a sensitivity analysis.
The proportion of individuals reporting any symptoms in the month prior to swabbing and any of the most predictive COVID-19 symptoms in the month prior to swabbing was calculated for the 8 most prevalent lineages across rounds 14 and 15. P-values were estimated for each lineage relative to AY.4 by performing logistic regression with the symptom status as a binary variable (any symptoms vs no symptoms, and separately most predictive COVID-19 symptoms vs none of the most predictive COVID-19 symptoms). The sensitivity of the results that AY.4.2 is less likely to exhibit the most predictive COVID-19 symptoms, relative to AY.4, was assessed by fitting further logistic regression models including age, round of study and N-gene Ct value as covariates (E-gene was also investigated but was no different to using N-gene and so this was not included).
Results
Lineage diversity.
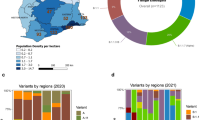
In round 14 the lineage was determined for 481 of 764 positive samples. All lineages were Delta or a Delta sub-lineage with the four most prevalent lineages being AY.4 at 65.1% (60.7%, 69.2%, n = 313), AY.43 at 6.0% (4.2%, 8.5%, n = 29), B.1.617.2 (original Delta variant) at 5.2% (3.6%, 7.6%, n = 25) and AY.4.2 at 4.6% (3.0%, 6.8%, n = 22) (Fig. 1-A, Additional file 2: Table S1). In round 15 the lineage was determined for 841 of 1399 positive samples. Again all samples were Delta or a Delta sub-lineage with the most prevalent lineages again being AY.4 at 57.6% (54.2%, 60.9%, n = 484), B.1.617.2 at 12.8% (10.8%, 15.3%, n = 108), AY.4.2 at 11.8% (9.8%, 14.1%, n = 99) and AY.43 at 4.8% (3.5%, 6.4%, n = 40). The next four most prevalent lineages over both rounds combined were AY.5, AY.6, AY.39, and AY.44. However, even a single detection of a lineage corresponded nationally to an average of 971 (95% CI [171, 5463]) individuals that would test swab-positive on any given day during round 14 and 1051 (95% CI [185, 5928]) individuals that would test swab-positive on any given day during round 15. During rounds 14 and 15 there were 33 and 31 unique lineages detected, respectively with 44 unique lineages detected overall. There was no apparent difference in genetic diversity between the two rounds as estimated by the Shannon diversity (p = 0.831) (Additional file 2: Table S2).

Proportion of positives by Delta sub-lineage. A The proportion of positives samples by round designated to the 8 lineages most prevalent over both rounds 14 and 15 (AY.39, AY.4, AY.4.2, AY.43, AY.44, AY.5, AY.6, B.1.617.2). B–D Proportion of positive samples by round and region with lineage designated as B B.1.617.2, C AY.4 and D AY.4.2. For all figures, point estimates of proportion are shown (bars) with 95% confidence intervals (error-bars)
Distribution by region and age
During round 15 the proportion of B.1.617.2 was found to be highest in London at 22.1% (14.9%, 31.4%), being greater than the proportion in South East, East of England and Yorkshire and The Humber (Fig. 1B, Additional file 2: Table S3). Conversely, in round 14 and 15 the proportion of AY.4 was lowest in London at 48.1% (35.4%, 61.1%) and 44.2% (34.6%, 54.2%) respectively and was found to be higher in North West, West Midlands and Yorkshire and The Humber during both rounds (Fig. 1C, Additional file 2: Table S3). This reduced proportion of the nationally most prevalent lineage (AY.4) in London coincided with a higher level of genetic diversity in London. The Shannon diversity was highest in London during both rounds at 1.814 in round 14 and 1.809 in round 15 (p < 0.001 and p = 0.002 respectively, reference = West Midlands, Additional file 2: Table S2). Higher levels of genetic diversity were also found during both rounds in the South East and South West, relative to the West Midlands (which showed the lowest levels of genetic diversity in round 14 and the second lowest in round 15). There were no regional differences in the proportion of AY.4.2 during round 15 (Fig. 1D, Additional file 2: Table S3). Regional differences during round 14 and regional differences for other lineages could not be investigated due to small sample sizes but numbers are provided in Additional file 2: Table S4.
Sub-regional analysis was performed in order to investigate the presence of clustering in each round for each lineage (see Methods). Despite being highly geographically dispersed (Fig. 2) clustering was detected in round 14 for AY.4 (p = 0.037) and AY.4.2 (p = 0.029) (Additional file 2: Table S5). However, during round 15 clustering was no longer evident for both AY.4 (p = 0.706) and AY.4.2 (p = 0.067). The only lineage for which clustering was detected in round 15 was AY.6 (p = 0.003) which was found mainly in London and towards the South coast of England.

Geographic distribution of all positive samples with a lineage designation (Green) with overlaid distribution of AY.4.2 (Pink, left), AY.4 (Purple, centre) and AY.6 (Orange, right) for both round 14 (top) and round 15 (bottom). The lineages shown had either a significant level of clustering in round 14 (AY.4 and AY.4.2) or round 15 (AY.6)
During round 15 the proportion of B.1.617.2 was higher in individuals ages 25–34 years old at 24.2% (12.8%, 41.0%) relative to those aged 35–44 years old at 8.0% (4.1%, 15.0%) (p = 0.026) (Additional file 2: Table S6). The proportion of AY.4 was found to be lower in 5–12 year olds at 52.1% (44.6%, 59.5%) relative to 35–44 year olds in which the proportion of AY.4 was 65.0% (55.3%, 73.6%) (p = 0.042) in round 15, while it was not in round 14.There were no differences between age groups in the proportion of AY.4.2 during round 15. Differences between age groups during round 14 for AY.4.2 and other lineages could not be investigated due to small sample sizes but numbers are provided in Additional file 2: Table S7.
Detection of increasing sub-lineages
Logistic regression models were fitted to the proportion of each lineage detected in either round 14 or 15, allowing daily growth rates in proportion to be estimated (Fig. 3, Additional file 2: Table S8). Of the 44 unique lineages detected, 6 were estimated to have growth rates different to zero. AY.4, AY.39, AY.98.1 and AY.111 were decreasing in proportion, whereas AY.4.2 and B.1.617.2 were increasing in proportion. The decrease in proportion of AY.4 corresponded to a daily growth rate of − 0.009 (− 0.015, − 0.003). The increase in proportions of B.1.617.2 and AY.4.2 corresponded to growth rates of 0.029 (0.017, 0.041) and 0.028 (0.016, 0.041) respectively.

A Estimated daily growth rate of the log odds of each lineage detected relative to all other lineages. Shown are both lineages with a growth rate in proportion not significantly different to zero (black) and those with a growth rate in proportion significantly different to zero (coloured). B–G Raw estimates of the daily proportions (points) with 95% confidence intervals (error bars) for lineages with a growth rate in proportion significantly different to zero: B.1.617.2 (B, pink), AY.4.2 (C, yellow), AY.4 (D, dark green), AY.111 (E, orange), AY.39 (F, purple), AY.98.1 (G, light green). Also shown is the best-fit Bayesian logistic regression model with central estimate (solid line) and 95% credible interval (shaded region)
Comparing estimates of the reproduction number R from round 14 to round 15 for AY.4 and AY.4.2 (see Methods) we estimate a multiplicative R advantage of 1.15 (1.08, 1.23), assuming no change in the generation time distribution.
Differences in cycle threshold values
There were quantitative differences between lineages in the N- and E-gene Ct values. The mean N- and E-gene Ct values were lowest for AY.6 though not materially lower than the values obtained for AY.4 (Fig. 4, Additional file 2: Table S9). Mean N-gene Ct value was 22.14 (20.30, 23.99) for AY.6 compared to 23.98 (23.68, 24.28) for AY.4 (p = 0.054). Mean E-gene Ct value was 20.74 (18.90, 22.59) for AY.6 compared to 22.46 (22.16, 22.76) for AY.4 (p = 0.071). Mean N- and E-gene Ct values were found to be comparable to AY.4 for both AY.4.2 and AY.5. Relative to AY.4, mean N- and E-gene Ct values for AY.43, AY.44, AY.39 and B.1.617.2 were all higher.

Estimated mean N-gene (A) and E-gene (B) Ct values for the 8 lineages most prevalent over rounds 14 and 15 (AY.39, AY.4, AY.4.2, AY.43, AY.44, AY.5, AY.6 and B.1.617.2) as calculated using Gaussian regression. Point estimates (points) and 95% confidence intervals (lines) are shown for estimates obtained using data from both rounds (blue), data from just round 14 (green) and data from just round 15 (purple)
Differences in symptomatology
The proportion of individuals exhibiting the most predictive COVID-19 symptoms (loss or change of sense of taste, loss or change of sense of smell, new persistent cough, fever) in the month prior to swabbing was lower (p = 0.029) in those infected with AY.4.2 at 42.1% (33.1%, 51.5%) relative to those infected with AY.4 at 53.4% (49.7%, 57.1%) (Fig. 5A, Additional file 2: Table S10). This difference was not explained by patterns in age, round of the study or N-gene Ct value (Fig. 5B, Additional file 2: Table S11).

A Proportion of positive individuals reporting any symptoms or reporting one of the four most predictive COVID-19 symptoms (loss or change of sense of taste, loss or change of sense of smell, new persistent cough, fever) in the last month by lineage of infection, for the 8 lineages most prevalent during rounds 14 and 15 (AY.39, AY.4, AY.4.2, AY.43, AY.44, AY.5, AY.6 and B.1.617.2). Point estimates of proportion are shown (bars) with 95% confidence intervals (error-bars). B Odds ratios of reporting the most predictive COVID-19 symptoms in the last months for multivariable logistic regression models including lineage (AY.4.2 with reference AY.4, red), age (relative to change of 10 years in age, blue), round of study (round 15 with reference round 14, green) and N-gene Ct value (relative to change in Ct value of 5, purple). The central estimates of odds ratios are shown (points) with 95% confidence intervals (error-bars)
In addition, 68.6% (59.8%, 76.3%) of those infected with AY.4.2 reported any symptoms in the month prior to swabbing compared to 75.4% (72.2%, 78.3%) for those infected with AY.4 (p = 0.119). There were no differences evident in symptom reporting between AY.4 infected individuals and the other 6 most prevalent lineages (B.1.617.2, AY.5, AY.6, AY.43, AY.44 and AY.39).
Phylogeographic analysis
A relaxed molecular clock model was fit to the data and used to estimate a time-resolved phylogenetic tree (Fig. 6). AY.4.2 was found to populate two closely related clades that emerged in June/July 2021. AY.43, AY.5 and AY.6 were also observed to have distinct clade groupings having emerged around June/July 2021 as well. The mutation rates inferred at the tree’s tips showed a large degree of variation in all of the 8 most prevalent lineages. The mean mutation rate for AY.4.2 was found to be 0.57 (< 0.01, 1.10)\(\times 1{0}^{-4}\) lower than the mean mutation rate of AY.4 (p = 0.050) (Fig. 6, Additional file 2: Table S2). The mean mutation rate inferred for samples collected in round 15 was found to be 1.00 (0.70, 1.40)\(\times 1{0}^{-4}\) lower than the mean mutation rate for samples collected in round 14 (p < 0.001).

A Time-resolved phylogenetic tree of all positive samples obtained for which the lineage designated was Delta or a Delta sub-lineage. B Distribution of mutation rates inferred at each phylogenetic tree tip for the 8 lineages most prevalent in round 14 and round 15 for samples obtained in round 14 and 15. C Distribution of mutation rates inferred at each phylogenetic tree tip for samples collected in round 14 and round 15. In B and C the estimates for mutation rate at each tip are given (points), the median (horizontal line) and interquartile range (boxes) of each distribution, and the range of the maximum and minimum values that are within 1.5 times the interquartile range of the box (vertical line). D Difference in the mean mutation rates between lineages and rounds as inferred from a Gaussian regression model. Central estimates of the differences are shown (points) with 95% confidence intervals (error-bars)
A mugration model was run on the time-resolved phylogenetic tree to estimate the relative virus migration rates between regions, a measure of inter-region transmission (Additional file 2: Table S13). Overall levels of inter-region transmission were lowest for the North East during round 14 and 15. The highest overall level of inter-region transmission was observed for the North West during round 14 and 15, but looking at individual rounds there were higher levels for Yorkshire and The Humber in round 14 and for the South East in round 15. High rates of transmission during round 14 and 15 were found between the North West and Yorkshire and The Humber, the West Midlands and the South East, and also between the South East and London.
Discussion
The proportion of AY.4.2 was found to be increasing between 9 September and 5 November 2021, as also reported in the routine data surveillance for England [11]. In round 15, AY.4.2 represented 11.8% of infections in line with other estimates [11]. This increase in proportion corresponded to a 15% increase in transmission advantage although this assumes the generation time distribution has remained constant; a decrease of the generation time distribution for AY.4.2 would also explain the increased growth but we are unable to test for this with prevalence data. In the past, the A222V mutation, associated with AY.4.2, increased in frequency but this was eventually deemed to be due to a founder effect and not a transmission advantage [40, 41]. Given the high levels of geographic dispersion (though with some clustering) during rounds 14 and 15 it is highly unlikely that a founder effect can explain the current growth, though we can not rule out a similar effect due to higher proportions of AY.4.2 in school-aged children (prevalence increased to a greater extent in school-aged children than in adults from July to September 2021 [4, 42]). However, as the proportion AY.4.2 was approximately constant by age in round 15 this growth advantage would not be detected into the future if this was the case.
Observed distributions of N- and E-gene Ct values were similar in AY.4.2 and AY.4 and so it is unlikely that the transmission advantage observed can be attributed to a higher viral load (a Ct 1 unit lower corresponds to an approximate twofold increase in viral load [43]). However, a reduced proportion of AY.4.2 infected individuals reporting symptoms could explain the increased transmissibility in multiple ways. Higher levels of asymptomatic infection could lead to greater levels of asymptomatic transmission. Further current testing procedures and government isolation advice in England heavily focus on the most predictive COVID-19 symptoms, which are reported less often by AY.4.2 infected individuals compared with AY.4. Thus, symptom-based policies could introduce an advantage for AY.4.2 over AY.4. Finally, the reduced level of symptom reporting could be indicative of greater levels of re-infection if AY.4.2 were more successful at evading the immune response. However, studies have found that vaccines are no less effective against AY.4.2 than other Delta sub-lineages [11] and vaccine-induced antibody neutralisation titres for AY.4.2 are similar to those for AY.4 and B.1.617.2 [44]. However, any possible evasion of the immune response caused by natural infections has yet to be investigated and the numbers reporting previous infection is too small and the proportion vaccinated too large in this REACT-1 dataset to allow a meaningful comparison (715 of 817 [87.5%] individuals aged 18 and over reported having had two vaccine doses). We found a moderately reduced mutation rate of AY.4.2 relative to AY.4 which may also have introduced a fitness advantage due to a smaller number of deleterious mutations [45, 46].
Other lineages
Though we have focused on AY.4.2 we have detected a diverse set of Delta sub-lineages, with even a single detection corresponding to approximately 1000 swab-positive infections in the community at one time during the study period. The short time over which AY.4.2 went from being an undeclared lineage to a variant under investigation shows how crucial it is to have careful surveillance of all lineages irrespective of frequency. For 38 of the 44 detected lineages, it was unable to be determined whether the proportion was increasing or decreasing.
Between rounds 14 and 15 a reduction in the mean mutation rate of the virus was detected suggesting a reduction in the rate of evolution. However, despite this slowdown evolution is still occurring and we observed an increase in the proportion of B.1.617.2, an indicator that the number of undeclared B.1.617.2 sub-lineages was increasing, suggesting even further diversity of Delta sub-lineages that have yet to be given a unique lineage designation. Further, though we capture the dynamics within England, SARS-CoV-2 is a global problem and new variants of concern can arise anywhere in the world and then spread through international travel. Higher proportions of B.1.617.2 were detected in London as well as higher levels of diversity; this likely reflects the role London continues to play in the introduction of international variants [47]. Within England, the North West region played a major role in the dissemination of the virus, having the greatest inferred rate of inter-region transmission.
Analysis of N- and E-gene Ct values found decreased levels in AY.4 and AY.4.2, which is unsurprising given both have successfully disseminated across the country, but AY.5 and AY.6 were also found to have similarly low Ct values suggesting similar viral loads; the mean N- and E-gene Ct value appeared slightly lower for AY.6 compared to AY.4. Clustering was also detected in round 15 for AY.6; careful consideration of AY.6 should be given in the future in case the current lack of growth so far reported [11] has only been due to its geographic isolation.
Limitations
We have presented the inferred dynamics between Delta sub-lineages in England between 9 September and 5 November 2021. Our sample's main strength over those obtained from routine surveillance is the random nature of the testing program leading to a relatively unbiased set of positive samples. However, as the sample sizes we obtain are relatively small compared with routine national surveillance our estimates have lower precision. Lineages were only successfully determined for ~ 61% of positive samples, with the ability to determine a lineage heavily influenced by a sample’s Ct value; this has potentially led to biases with lineages with lower Ct values more heavily represented in the dataset. Detecting distinct sub-lineages is a high-dimensional problem, with often many common mutations being shared between distinct lineages with only a small number of distinguishing mutations. This is exacerbated when all the lineages are highly related, as in the current nature of the pandemic in England where all samples are descendants of Delta (B.1.617.2), and can lead to incorrect designations [48]. Further, only sub-lineages that have been defined are able to be assigned to a sample. During the emergence of a new sub-lineage there is a phase of ambiguity when numbers are small and it is unclear if the mutations present warrant the declaration of a new sub-lineage. This can be seen in the detection of AY.4.2 and AY.43; both lineages had been circulating for months by October 2021 [11] but were not yet declared sub-lineages by pangoLEARN [28] in early October 2021, and so did not appear in the publicly available technical briefings [49]. The analysis of mutation rates using Gaussian regression may also have included biases as individual measurements of mutation rates would not have independent and identically distributed normal errors, a key assumption of these linear models.
Conclusions
Since the beginning of the pandemic, selective pressure has led to rapid evolution in the spike protein [50] driving leaps in transmissibility [5]. However, as a greater proportion of the population acquires immunity through either infection or vaccination there will be a shift in evolutionary pressure towards immune escape. Even in England where there are high levels of vaccination and past infection, new variants such as AY.4.2 have emerged with advantages over previous strains. With the continued emergence of variants able to evade population immunity and undergoing transmission, SARS-CoV-2 is highly unlikely to ever undergo local extinction and is likely moving towards a state of endemicity. At the point of endemicity it is probable that adaptive evolution would more closely resemble the continual antigenic drift observed in influenza H3N2 [51, 52]. As the evolutionary phase of SARS-CoV-2 progresses towards endemicity, continued surveillance is paramount in not only detecting increased levels of transmissibility for specific lineages, but in also better characterising the mechanism behind such changes and informing policy around testing (including case definitions). Representative community studies such as REACT-1 can be useful in measuring the relative growth of lineages and in characterising differences in viral loads, symptomatology and geographic distribution.
Availability of data and materials
Access to REACT-1 data is restricted due to ethical and security considerations. Summary statistics and descriptive tables from the current REACT-1 study are available in the Additional file 2. Additional summary statistics and results from the REACT-1 programme are also available at https://www.imperial.ac.uk/medicine/research-and-impact/groups/react-study/real-time-assessment-of-community-transmission-findings/ and https://github.com/mrc-ide/reactidd/tree/master/inst/extdata REACT-1 Study Materials are available for each round at https://www.imperial.ac.uk/medicine/research-and-impact/groups/react-study/react-1-study-materials/. Sequence read data are available without restriction from the European Nucleotide Archive at https://www.ebi.ac.uk/ena/browser/view/PRJEB37886, and consensus genome sequences are available from the Global initiative on sharing all influenza data at https://www.gisaid.org. Accession numbers are provided in the Additional file 1.
References
Lineage B.1.617.2 Pangolin report. https://cov-lineages.org/global_report_B.1.617.2.html. Accessed 26 Nov 2021.
World Health Organisation. COVID-19 Weekly Epidemiological Update - Edition 66. 2021. https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---16-november-2021. Accessed 23 Nov 2021.
PHE Genomics Cell, PHE Outbreak Surveillance Team, PHE Epidemiology Cell, PHE Contact Tracing Data Team, PHE Health, Protection Data Science Team, PHE Joint Modelling Team, NHS Test and Trace Joint Biosecurity Centre, Public Health Scotland and EAVE group, Contributions from the Variant Technical Group Members. SARS-CoV-2 variants of concern and variants under investigation in England - Technical briefing 15, 11 June 2021. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/993879/Variants_of_Concern_VOC_Technical_Briefing_15.pdf
Elliott P, Haw D, Wang H, Eales O, Walters CE, Ainslie KEC, et al. Exponential growth, high prevalence of SARS-CoV-2, and vaccine effectiveness associated with the Delta variant. Science. 2021;374:eabl9551.
Obermeyer F, Schaffner SF, Jankowiak M, Barkas N, Pyle JD, Park DJ, et al. Analysis of 2.1 million SARS-CoV-2 genomes identifies mutations associated with transmissibility. https://doi.org/10.1101/2021.09.07.21263228
Bernal JL, Andrews N, Gower C, Gallagher E, Simmons R, Thelwall S, et al. Effectiveness of COVID-19 vaccines against the B.1.617.2 variant. medRxiv. 2021; 2021.05.22.21257658.
Baj A, Novazzi F, Drago Ferrante F, Genoni A, Tettamanzi E, Catanoso G, et al. Spike protein evolution in the SARS-CoV-2 Delta variant of concern: a case series from Northern Lombardy. Emerg Microbes Infect. 2021;10:2010–5.
Kistler KE, Huddleston J, Bedford T. Rapid and parallel adaptive mutations in spike S1 drive clade success in SARS-CoV-2. BioRxiv. 2021. https://doi.org/10.1101/2021.09.11.459844.
Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5:1403–7.
UKHSA Genomics Cell UKHSA Outbreak Surveillance Team UKHSA Epidemiology Cell UKHSA Contact Tracing Data Team UKHSA International Cell UKHSA Environmental Monitoring for Health Protection Team. SARS-CoV-2 variants of concern and variants under investigation in England - Technical briefing 27, 29 October 2021. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1029715/technical-briefing-27.pdf
UKHSA Genomics Cell UKHSA Outbreak Surveillance Team UKHSA Epidemiology Cell UKHSA Contact Tracing Data Team UKHSA International Cell UKHSA Environmental Monitoring for Health Protection Team. SARS-CoV-2 variants of concern and variants under investigation in England - Technical briefing 28, 12 November 2021. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1033101/Technical_Briefing_28_12_Nov_2021.pdf
UKHSA Genomics Cell UKHSA Outbreak Surveillance Team UKHSA Epidemiology Cell UKHSA Contact Tracing Data Team UKHSA International Cell UKHSA Environmental Monitoring for Health Protection Team. SARS-CoV-2 variants of concern and variants under investigation in England - Technical briefing 26, 22 October 2021. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1028113/Technical_Briefing_26.pdf
Lineage AY.4.2 Pangolin report. https://cov-lineages.org/lineage.html?lineage=AY.4.2. Accessed 23 Nov 2021.
Alaa Abdel Latif, Julia L. Mullen, Manar Alkuzweny, Ginger Tsueng, Marco Cano, Emily Haag, Jerry Zhou, Mark Zeller, Emory Hufbauer, Nate Matteson, Chunlei Wu, Kristian G. Andersen, Andrew I. Su, Karthik Gangavarapu, Laura D. Hughes, and the Center for Viral Systems Biology. AY.4 lineage report, outbreak.info. https://outbreak.info/situation-reports?pango=AY.4.2. Accessed 23 Nov 2021.
Lineage AY.4 Pangolin report. https://cov-lineages.org/lineage.html?lineage=AY.4. Accessed 23 Nov 2021.
Umair M, Ikram A, Rehman Z, Haider A, Badar N, Ammar M, et al. Genomic diversity of SARS-CoV-2 in Pakistan during fourth wave of pandemic. BioRxiv. 2021. https://doi.org/10.1101/2021.09.30.21264343.
Danish Covid-19 Genome Consortium. Genomic overview of SARS-CoV-2 in Denmark, 19 November 2021. https://www.covid19genomics.dk/statistics. Accessed 23 Nov 2021.
Official UK Coronavirus Dashboard. https://coronavirus.data.gov.uk/. Accessed 17 May 2021.
Latest insights team. Coronavirus (COVID-19) latest insights - Office for National Statistics. Office for National Statistics; 23 Nov 2021. https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/articles/coronaviruscovid19latestinsights/antibodies. Accessed 25 Nov 2021.
Riley S, Ainslie KEC, Eales O, Walters CE, Wang H, Atchison C, et al. Resurgence of SARS-CoV-2: detection by community viral surveillance. Science. 2021. https://doi.org/10.1126/science.abf0874.
Ricoca Peixoto V, Nunes C, Abrantes A. Epidemic surveillance of Covid-19: considering uncertainty and under-ascertainment. Portuguese J Public Health. 2020;38:23–9.
Feng S, Phillips DJ, White T, Sayal H, Aley PK, Bibi S, et al. Correlates of protection against symptomatic and asymptomatic SARS-CoV-2 infection. Nat Med. 2021;27:2032–40.
Riley S, Atchison C, Ashby D, Donnelly CA, Barclay W, Cooke G, et al. REal-time Assessment of Community Transmission (REACT) of SARS-CoV-2 virus: study protocol. Wellcome Open Res. 2020;5:200.
Quick J. nCoV-2019 sequencing protocol v3 (LoCost). 2020. https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye. Accessed 4 May 2021.
Baker DJ, Aydin A, Le-Viet T, Kay GL, Rudder S, de Oliveira ML, et al. CoronaHiT: high-throughput sequencing of SARS-CoV-2 genomes. Genome Med. 2021;13:21.
A Nextflow pipeline for running the ARTIC network’s field bioinformatics tools. Github. https://github.com/connor-lab/ncov2019-artic-nf
Connor TR, Loman NJ, Thompson S, Smith A, Southgate J, Poplawski R, et al. CLIMB (the Cloud Infrastructure for Microbial Bioinformatics): an online resource for the medical microbiology community. Microb Genom. 2016;2: e000086.
Phylogenetic Assignment of Named Global Outbreak LINeages (PANGOLIN). Github; https://github.com/cov-lineages/pangolin
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Sagulenko P, Puller V, Neher RA. TreeTime: maximum-likelihood phylodynamic analysis. Virus Evol. 2018;4:vex042.
Wilson EB. Probable inference, the law of succession, and statistical inference. J Am Stat Assoc. 1927;22:209–12.
Brown LD, Tony Cai T, DasGupta A. Interval estimation for a binomial proportion. SSO Schweiz Monatsschr Zahnheilkd. 2001;16:101–33.
Chadeau-Hyam M, Eales O, Bodinier B, Wang H, Haw D, Whitaker M, et al. REACT-1 round 15 final report: Increased breakthrough SARS-CoV-2 infections among adults who had received two doses of vaccine, but booster doses and first doses in children are providing important protection. 2021. http://spiral.imperial.ac.uk/handle/10044/1/92501. Accessed 26 Nov 2021.
Park N. Population estimates for the UK, England and Wales, Scotland and Northern Ireland - Office for National Statistics. Office for National Statistics; 24 Jun 2021. https://www.ons.gov.uk/peoplepopulationandcommunity/populationandmigration/populationestimates/bulletins/annualmidyearpopulationestimates/mid2020. Accessed 11 Aug 2021.
Spellerberg IF, Fedor PJ. A tribute to Claude Shannon (1916–2001) and a plea for more rigorous use of species richness, species diversity and the “Shannon-Wiener” Index. Glob Ecol Biogeogr. 2003;12:177–9.
Hutcheson K. A test for comparing diversities based on the Shannon formula. J Theor Biol. 1970;29:151–4.
Sharot T. Weighting survey results. J Mark Res Soc. 1986;28:269–84.
Bi Q, Wu Y, Mei S, Ye C, Zou X, Zhang Z, et al. Epidemiology and Transmission of COVID-19 in Shenzhen China: Analysis of 391 cases and 1,286 of their close contacts. MedRxiv. 2020. https://www.medrxiv.org/content/medrxiv/early/2020/03/19/2020.03.03.20028423.full.pdf
Wallinga J, Lipsitch M. How generation intervals shape the relationship between growth rates and reproductive numbers. Proc Biol Sci. 2007;274:599–604.
Díez-Fuertes F, Iglesias-Caballero M, García-Pérez J, Monzón S, Jiménez P, Varona S, et al. A founder effect led early SARS-CoV-2 transmission in Spain. J Virol. 2021. https://doi.org/10.1128/JVI.01583-20.
Hodcroft EB, Zuber M, Nadeau S, Vaughan TG, Crawford KHD, Althaus CL, et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature. 2021;595:707–12.
Chadeau-Hyam M, Wang H, Eales O, Haw D, Bodinier B, Whitaker M, et al. REACT-1 study round 14: High and increasing prevalence of SARS-CoV-2 infection among school-aged children during September 2021 and vaccine effectiveness against infection in England. medRxiv. 2021; 2021.10.14.21264965.
Yelin I, Aharony N, Tamar ES, Argoetti A, Messer E, Berenbaum D, et al. Evaluation of COVID-19 RT-qPCR Test in Multi sample Pools. Clin Infect Dis. 2020;71:2073–8.
Lassaunière R, Polacek C, Fonager J, Bennedbæk M, Boding L, Rasmussen M, et al. Neutralisation of the SARS-CoV-2 Delta sub-lineage AY.4.2 and B.1.617.2 + E484K by BNT162b2 mRNA vaccine-elicited sera. bioRxiv. 2021. https://doi.org/10.1101/2021.11.08.21266075.
Peck KM, Lauring AS. Complexities of Viral Mutation Rates. J Virol. 2018. https://doi.org/10.1128/JVI.01031-17.
Koelle K, Rasmussen DA. The effects of a deleterious mutation load on patterns of influenza A/H3N2’s antigenic evolution in humans. Elife. 2015;4: e07361.
Eales O, Page AJ, Tang SN, Walters CE, Wang H, Haw D, et al. SARS-CoV-2 lineage dynamics in England from January to March 2021 inferred from representative community samples. medRxiv. 2021. https://doi.org/10.1101/2021.05.08.21256867.
O’Toole Á, Scher E, Underwood A, Jackson B, Hill V, McCrone JT, et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021;7:vea0b64.
UKHSA Genomics Cell UKHSA Outbreak Surveillance Team UKHSA Epidemiology Cell UKHSA Contact Tracing Data Team UKHSA International Cell UKHSA Environmental Monitoring for Health Protection Team. SARS-CoV-2 variants of concern and variants under investigation in England - Technical briefing 25, 1 October 2021. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1025827/Technical_Briefing_25.pdf
Saputri DS, Li S, van Eerden FJ, Rozewicki J, Xu Z, Ismanto HS, et al. Flexible, functional, and familiar: characteristics of SARS-CoV-2 spike protein evolution. Front Microbiol. 2020;11:2112.
Bedford T, Riley S, Barr IG, Broor S, Chadha M, Cox NJ, et al. Global circulation patterns of seasonal influenza viruses vary with antigenic drift. Nature. 2015;523:217–20.
Andrew Rambaut, Oliver G. Pybus, Martha I. Nelson, Cecile Viboud, Jeffery K. Taubenberger, Edward C. Holmes. The genomic and epidemiological dynamics of human influenza A virus. Nature. 2008. https://www.nature.com/articles/nature06945
Acknowledgements
MC-H acknowledges support from the H2020-EXPANSE project (Horizon 2020 Grant No 874627). MC-H and BB acknowledge support from Cancer Research UK, Population Research Committee Project grant 'Mechanomics’ (Grant No 22184 to MC-H). CAD acknowledges support from the MRC Centre for Global Infectious Disease Analysis and National Institute for Health Research (NIHR) Health Protection Research Unit (HPRU). GC is supported by an NIHR Professorship. HW acknowledges support from an NIHR Senior Investigator Award and the Wellcome Trust (205456/Z/16/Z). PE is Director of the Medical Research Council (MRC) Centre for Environment and Health (MR/L01341X/1, MR/S019669/1). PE acknowledges support from Health Data Research UK (HDR UK); the NIHR Imperial Biomedical Research Centre; NIHR Health Protection Research Units in Chemical and Radiation Threats and Hazards, and Environmental Exposures and Health; the British Heart Foundation Centre for Research Excellence at Imperial College London (RE/18/4/34215); and the UK Dementia Research Institute at Imperial College London (MC_PC_17114). AJP acknowledges the support of the Biotechnology and Biological Sciences Research Council (BB/R012504/1). We thank The Huo Family Foundation for their support of our work on COVID-19.
We thank key collaborators on this work—Ipsos MORI: Kelly Beaver, Sam Clemens, Gary Welch, Nicholas Gilby, Kelly Ward, Galini Pantelidou and Kevin Pickering; Institute of Global Health Innovation at Imperial College London: Gianluca Fontana, Justine Alford; School of Public Health, Imperial College London: Eric Johnson, Rob Elliott, Graham Blakoe; Quadram Institute, Norwich, UK: Alexander J. Trotter; North West London Pathology and Public Health England (now UKHSA) for help in calibration of the laboratory analyses; Patient Experience Research Centre at Imperial College London and the REACT Public Advisory Panel; NHS Digital for access to the NHS register; the Department of Health and Social Care for logistic support.
The COVID-19 Genomics UK (COG-UK) consortium—June 2021 V.1
Funding acquisition, Leadership and supervision, Metadata curation, Project administration, Samples and logistics, Sequencing and analysis, Software and analysis tools, and Visualisation:
Dr Samuel C Robson13,84
Funding acquisition, Leadership and supervision, Metadata curation, Project administration, Samples and logistics, Sequencing and analysis, and Software and analysis tools:
Dr Thomas R Connor11,74 and Prof Nicholas J Loman43
Leadership and supervision, Metadata curation, Project administration, Samples and logistics, Sequencing and analysis, Software and analysis tools, and Visualisation:
Dr Tanya Golubchik5
Funding acquisition, Leadership and supervision, Metadata curation, Samples and logistics, Sequencing and analysis, and Visualisation:
Dr Rocio T Martinez Nunez46
Funding acquisition, Leadership and supervision, Project administration, Samples and logistics, Sequencing and analysis, and Software and analysis tools:
Dr David Bonsall5
Funding acquisition, Leadership and supervision, Project administration, Sequencing and analysis, Software and analysis tools, and Visualisation:
Prof Andrew Rambaut104
Funding acquisition, Metadata curation, Project administration, Samples and logistics, Sequencing and analysis, and Software and analysis tools:
Dr Luke B Snell12
Leadership and supervision, Metadata curation, Project administration, Samples and logistics, Software and analysis tools, and Visualisation:
Rich Livett116
Funding acquisition, Leadership and supervision, Metadata curation, Project administration, and Samples and logistics:
Dr Catherine Ludden20,70
Funding acquisition, Leadership and supervision, Metadata curation, Samples and logistics, and Sequencing and analysis:
Dr Sally Corden74 and Dr Eleni Nastouli96,95,30
Funding acquisition, Leadership and supervision, Metadata curation, Sequencing and analysis, and Software and analysis tools:
Dr Gaia Nebbia12
Funding acquisition, Leadership and supervision, Project administration, Samples and logistics, and Sequencing and analysis:
Ian Johnston116
Leadership and supervision, Metadata curation, Project administration, Samples and logistics, and Sequencing and analysis:
Prof Katrina Lythgoe5, Dr M. Estee Torok19,20 and Prof Ian G Goodfellow24
Leadership and supervision, Metadata curation, Project administration, Samples and logistics, and Visualisation:
Dr Jacqui A Prieto97,82 and Dr Kordo Saeed97,83
Leadership and supervision, Metadata curation, Project administration, Sequencing and analysis, and Software and analysis tools:
Dr David K Jackson116
Leadership and supervision, Metadata curation, Samples and logistics, Sequencing and analysis, and Visualisation:
Dr Catherine Houlihan96,94
Leadership and supervision, Metadata curation, Sequencing and analysis, Software and analysis tools, and Visualisation:
Dr Dan Frampton94,95
Metadata curation, Project administration, Samples and logistics, Sequencing and analysis, and Software and analysis tools:
Dr William L Hamilton19 and Dr Adam A Witney41
Funding acquisition, Samples and logistics, Sequencing and analysis, and Visualisation:
Dr Giselda Bucca101
Funding acquisition, Leadership and supervision, Metadata curation, and Project administration:
Dr Cassie F Pope40,41
Funding acquisition, Leadership and supervision, Metadata curation, and Samples and logistics:
Dr Catherine Moore74
Funding acquisition, Leadership and supervision, Metadata curation, and Sequencing and analysis:
Prof Emma C Thomson53
Funding acquisition, Leadership and supervision, Project administration, and Samples and logistics:
Dr Ewan M Harrison116,102
Funding acquisition, Leadership and supervision, Sequencing and analysis, and Visualisation:
Prof Colin P Smith101
Leadership and supervision, Metadata curation, Project administration, and Sequencing and analysis:
Fiona Rogan77
Leadership and supervision, Metadata curation, Project administration, and Samples and logistics:
Shaun M Beckwith 6, Abigail Murray 6, Dawn Singleton 6, Dr Kirstine Eastick 37, Dr Liz A Sheridan 98, Paul Randell 99, Dr Leigh M Jackson 105, Dr Cristina V Ariani 116 and Dr Sónia Gonçalves116
Leadership and supervision, Metadata curation, Samples and logistics, and Sequencing and analysis:
Dr Derek J Fairley 3,77, Prof Matthew W Loose 18 and Joanne Watkins74
Leadership and supervision, Metadata curation, Samples and logistics, and Visualisation:
Dr Samuel Moses25,106
Leadership and supervision, Metadata curation, Sequencing and analysis, and Software and analysis tools:
Dr Sam Nicholls 43, Dr Matthew Bull 74 and Dr Roberto Amato116
Leadership and supervision, Project administration, Samples and logistics, and Sequencing and analysis:
Prof Darren L Smith36,65,66
Leadership and supervision, Sequencing and analysis, Software and analysis tools, and Visualisation:
Prof David M Aanensen14,116 and Dr Jeffrey C Barrett116
Metadata curation, Project administration, Samples and logistics, and Sequencing and analysis:
Dr Dinesh Aggarwal20,116,70, Dr James G Shepherd 53, Dr Martin D Curran 71 and Dr Surendra Parmar71
Metadata curation, Project administration, Sequencing and analysis, and Software and analysis tools:
Dr Matthew D Parker109
Metadata curation, Samples and logistics, Sequencing and analysis, and Software and analysis tools:
Dr Catryn Williams74
Metadata curation, Samples and logistics, Sequencing and analysis, and Visualisation:
Dr Sharon Glaysher68
Metadata curation, Sequencing and analysis, Software and analysis tools, and Visualisation:
Dr Anthony P Underwood 14,116, Dr Matthew Bashton 36,65, Dr Nicole Pacchiarini 74, Dr Katie F Loveson84 and Matthew Byott95,96
Project administration, Sequencing and analysis, Software and analysis tools, and Visualisation:
Dr Alessandro M Carabelli20
Funding acquisition, Leadership and supervision, and Metadata curation:
Dr Kate E Templeton56,104
Funding acquisition, Leadership and supervision, and Project administration:
Dr Thushan I de Silva109, Dr Dennis Wang109, Dr Cordelia F Langford116 and John Sillitoe116
Funding acquisition, Leadership and supervision, and Samples and logistics:
Prof Rory N Gunson55
Funding acquisition, Leadership and supervision, and Sequencing and analysis:
Dr Simon Cottrell74, Dr Justin O’Grady75,103 and Prof Dominic Kwiatkowski116,108
Leadership and supervision, Metadata curation, and Project administration:
Dr Patrick J Lillie37
Leadership and supervision, Metadata curation, and Samples and logistics:
Dr Nicholas Cortes33, Dr Nathan Moore33, Dr Claire Thomas33, Phillipa J Burns37, Dr Tabitha W Mahungu80 and Steven Liggett86
Leadership and supervision, Metadata curation, and Sequencing and analysis:
Angela H Beckett13,81 and Prof Matthew TG Holden73
Leadership and supervision, Project administration, and Samples and logistics:
Dr Lisa J Levett34, Dr Husam Osman70,35 and Dr Mohammed O Hassan-Ibrahim99
Leadership and supervision, Project administration, and Sequencing and analysis:
Dr David A Simpson77
Leadership and supervision, Samples and logistics, and Sequencing and analysis:
Dr Meera Chand72, Prof Ravi K Gupta102, Prof Alistair C Darby107 and Prof Steve Paterson107
Leadership and supervision, Sequencing and analysis, and Software and analysis tools:
Prof Oliver G Pybus23, Dr Erik M Volz39, Prof Daniela de Angelis52, Prof David L Robertson53, Dr Andrew J Page75 and Dr Inigo Martincorena116
Leadership and supervision, Sequencing and analysis, and Visualisation:
Dr Louise Aigrain116 and Dr Andrew R Bassett116
Metadata curation, Project administration, and Samples and logistics:
Dr Nick Wong50, Dr Yusri Taha89, Michelle J Erkiert99 and Dr Michael H Spencer Chapman116,102
Metadata curation, Project administration, and Sequencing and analysis:
Dr Rebecca Dewar56 and Martin P McHugh56,111
Metadata curation, Project administration, and Software and analysis tools:
Siddharth Mookerjee38,57
Metadata curation, Project administration, and Visualisation:
Stephen Aplin97, Matthew Harvey97, Thea Sass97, Dr Helen Umpleby97 and Helen Wheeler97
Metadata curation, Samples and logistics, and Sequencing and analysis:
Dr James P McKenna3, Dr Ben Warne9, Joshua F Taylor22, Yasmin Chaudhry24, Rhys Izuagbe24, Dr Aminu S Jahun24, Dr Gregory R Young 6,65, Dr Claire McMurray43, Dr Clare M McCann65,66, Dr Andrew Nelson65,66 and Scott Elliott68
Metadata curation, Samples and logistics, and Visualisation:
Hannah Lowe25
Metadata curation, Sequencing and analysis, and Software and analysis tools:
Dr Anna Price11, Matthew R Crown65, Dr Sara Rey74, Dr Sunando Roy96 and Dr Ben Temperton105
Metadata curation, Sequencing and analysis, and Visualisation:
Dr Sharif Shaaban73 and Dr Andrew R Hesketh101
Project administration, Samples and logistics, and Sequencing and analysis:
Dr Kenneth G Laing41, Dr Irene M Monahan41 and Dr Judith Heaney95,96,34
Project administration, Samples and logistics, and Visualisation:
Dr Emanuela Pelosi97, Siona Silviera97 and Dr Eleri Wilson-Davies97
Samples and logistics, Software and analysis tools, and Visualisation:
Dr Helen Fryer5
Sequencing and analysis, Software and analysis tools, and Visualization:
Dr Helen Adams4, Dr Louis du Plessis23, Dr Rob Johnson39, Dr William T Harvey53,42, Dr Joseph Hughes53, Dr Richard J Orton53, Dr Lewis G Spurgin59, Dr Yann Bourgeois81, Dr Chris Ruis102, Áine O'Toole104, Marina Gourtovaia116 and Dr Theo Sanderson116
Funding acquisition, and Leadership and supervision:
Dr Christophe Fraser5, Dr Jonathan Edgeworth12, Prof Judith Breuer96,29, Dr Stephen L Michell105 and Prof John A Todd115
Funding acquisition, and Project administration:
Michaela John10 and Dr David Buck115
Leadership and supervision, and Metadata curation:
Dr Kavitha Gajee37 and Dr Gemma L Kay75
Leadership and supervision, and Project administration:
Prof Sharon J Peacock20,70 and David Heyburn74
Leadership and supervision, and Samples and logistics:
Katie Kitchman37, Prof Alan McNally43,93, David T Pritchard50, Dr Samir Dervisevic58, Dr Peter Muir70, Dr Esther Robinson70,35, Dr Barry B Vipond70, Newara A Ramadan78, Dr Christopher Jeanes90, Danni Weldon116, Jana Catalan118 and Neil Jones118
Leadership and supervision, and Sequencing and analysis:
Dr Ana da Silva Filipe53, Dr Chris Williams74, Marc Fuchs77, Dr Julia Miskelly77, Dr Aaron R Jeffries105, Karen Oliver116 and Dr Naomi R Park116
Metadata curation, and Samples and logistics:
Amy Ash1, Cherian Koshy1, Magdalena Barrow7, Dr Sarah L Buchan7, Dr Anna Mantzouratou7, Dr Gemma Clark15, Dr Christopher W Holmes16, Sharon Campbell17, Thomas Davis21, Ngee Keong Tan22, Dr Julianne R Brown29, Dr Kathryn A Harris29,2, Stephen P Kidd33, Dr Paul R Grant34, Dr Li Xu-McCrae35, Dr Alison Cox38,63, Pinglawathee Madona38,63, Dr Marcus Pond38,63, Dr Paul A Randell38,63, Karen T Withell48, Cheryl Williams 51, Dr Clive Graham60, Rebecca Denton-Smith62, Emma Swindells62, Robyn Turnbull62, Dr Tim J Sloan67, Dr Andrew Bosworth70,35, Stephanie Hutchings70, Hannah M Pymont70, Dr Anna Casey76, Dr Liz Ratcliffe76, Dr Christopher R Jones79,105, Dr Bridget A Knight79,105, Dr Tanzina Haque80, Dr Jennifer Hart80, Dr Dianne Irish-Tavares80, Eric Witele80, Craig Mower86, Louisa K Watson86, Jennifer Collins89, Gary Eltringham89, Dorian Crudgington98, Ben Macklin98, Prof Miren Iturriza-Gomara107, Dr Anita O Lucaci107 and Dr Patrick C McClure113
Metadata curation, and Sequencing and analysis:
Matthew Carlile18, Dr Nadine Holmes18, Dr Christopher Moore18, Dr Nathaniel Storey29, Dr Stefan Rooke73, Dr Gonzalo Yebra73, Dr Noel Craine74, Malorie Perry74, Dr Nabil-Fareed Alikhan75, Dr Stephen Bridgett77, Kate F Cook 84, Christopher Fearn84, Dr Salman Goudarzi84, Prof Ronan A Lyons88, Dr Thomas Williams104, Dr Sam T Haldenby107, Jillian Durham116 and Dr Steven Leonard116
Metadata curation, and Software and analysis tools:
Robert M Davies116
Project administration, and Samples and logistics:
Dr Rahul Batra12, Beth Blane20, Dr Moira J Spyer30,95,96, Perminder Smith32,112, Mehmet Yavus85,109, Dr Rachel J Williams96, Dr Adhyana IK Mahanama97, Dr Buddhini Samaraweera97, Sophia T Girgis102, Samantha E Hansford109, Dr Angie Green115, Dr Charlotte Beaver116, Katherine L Bellis116,102, Matthew J Dorman116, Sally Kay116, Liam Prestwood116 and Dr Shavanthi Rajatileka116
Project administration, and Sequencing and analysis:
Dr Joshua Quick43
Project administration, and Software and analysis tools:
Radoslaw Poplawski43
Samples and logistics, and Sequencing and analysis:
Dr Nicola Reynolds8, Andrew Mack11, Dr Arthur Morriss11, Thomas Whalley11, Bindi Patel12, Dr Iliana Georgana24, Dr Myra Hosmillo24, Malte L Pinckert24, Dr Joanne Stockton43, Dr John H Henderson65, Amy Hollis65, Dr William Stanley65, Dr Wen C Yew65, Dr Richard Myers72, Dr Alicia Thornton72, Alexander Adams74, Tara Annett74, Dr Hibo Asad74, Alec Birchley74, Jason Coombes74, Johnathan M Evans74, Laia Fina74, Bree Gatica-Wilcox74, Lauren Gilbert74, Lee Graham74, Jessica Hey74, Ember Hilvers74, Sophie Jones74, Hannah Jones74, Sara Kumziene-Summerhayes74, Dr Caoimhe McKerr74, Jessica Powell74, Georgia Pugh74, Sarah Taylor74, Alexander J Trotter75, Charlotte A Williams96, Leanne M Kermack102, Benjamin H Foulkes109, Marta Gallis109, Hailey R Hornsby109, Stavroula F Louka 109, Dr Manoj Pohare109, Paige Wolverson109, Peijun Zhang109, George MacIntyre-Cockett115, Amy Trebes115, Dr Robin J Moll116, Lynne Ferguson117, Dr Emily J Goldstein117, Dr Alasdair Maclean117 and Dr Rachael Tomb117
Samples and logistics, and Software and analysis tools:
Dr Igor Starinskij53
Sequencing and analysis, and Software and analysis tools:
Laura Thomson5, Joel Southgate11,74, Dr Moritz UG Kraemer23, Dr Jayna Raghwani23, Dr Alex E Zarebski23, Olivia Boyd39, Lily Geidelberg39, Dr Chris J Illingworth52, Dr Chris Jackson52, Dr David Pascall52, Dr Sreenu Vattipally53, Timothy M Freeman109, Dr Sharon N Hsu109, Dr Benjamin B Lindsey109, Dr Keith James116, Kevin Lewis116, Gerry Tonkin-Hill116 and Dr Jaime M Tovar-Corona116
Sequencing and analysis, and Visualisation:
MacGregor Cox20
Software and analysis tools, and Visualisation:
Dr Khalil Abudahab14,116, Mirko Menegazzo14, Ben EW Taylor MEng14,116, Dr Corin A Yeats14, Afrida Mukaddas53, Derek W Wright53, Dr Leonardo de Oliveira Martins75, Dr Rachel Colquhoun104, Verity Hill104, Dr Ben Jackson104, Dr JT McCrone104, Dr Nathan Medd104, Dr Emily Scher104 and Jon-Paul Keatley116
Leadership and supervision:
Dr Tanya Curran3, Dr Sian Morgan10, Prof Patrick Maxwell20, Prof Ken Smith20, Dr Sahar Eldirdiri21, Anita Kenyon21, Prof Alison H Holmes38,57, Dr James R Price38,57, Dr Tim Wyatt69, Dr Alison E Mather75, Dr Timofey Skvortsov77 and Prof John A Hartley96
Metadata curation:
Prof Martyn Guest11, Dr Christine Kitchen11, Dr Ian Merrick11, Robert Munn11, Dr Beatrice Bertolusso33, Dr Jessica Lynch33, Dr Gabrielle Vernet33, Stuart Kirk34, Dr Elizabeth Wastnedge56, Dr Rachael Stanley58, Giles Idle64, Dr Declan T Bradley69,77, Dr Jennifer Poyner79 and Matilde Mori110
Project administration:
Owen Jones11, Victoria Wright18, Ellena Brooks20, Carol M Churcher20, Mireille Fragakis20, Dr Katerina Galai20,70, Dr Andrew Jermy20, Sarah Judges20, Georgina M McManus20, Kim S Smith20, Dr Elaine Westwick20, Dr Stephen W Attwood23, Dr Frances Bolt38,57, Dr Alisha Davies74, Elen De Lacy74, Fatima Downing74, Sue Edwards74, Lizzie Meadows75, Sarah Jeremiah97, Dr Nikki Smith109 and Luke Foulser116
Samples and logistics:
Dr Themoula Charalampous12,46, Amita Patel12, Dr Louise Berry15, Dr Tim Boswell15, Dr Vicki M Fleming15, Dr Hannah C Howson-Wells15, Dr Amelia Joseph15, Manjinder Khakh15, Dr Michelle M Lister15, Paul W Bird16, Karlie Fallon16, Thomas Helmer16, Dr Claire L McMurray16, Mina Odedra16, Jessica Shaw16, Dr Julian W Tang16, Nicholas J Willford16, Victoria Blakey17, Dr Veena Raviprakash17, Nicola Sheriff17, Lesley-Anne Williams17, Theresa Feltwell20, Dr Luke Bedford26, Dr James S Cargill27, Warwick Hughes27, Dr Jonathan Moore28, Susanne Stonehouse28, Laura Atkinson29, Jack CD Lee29, Dr Divya Shah29, Adela Alcolea-Medina32,112, Natasha Ohemeng-Kumi32,112, John Ramble32,112, Jasveen Sehmi32,112, Dr Rebecca Williams33, Wendy Chatterton34, Monika Pusok34, William Everson37, Anibolina Castigador44, Emily Macnaughton44, Dr Kate El Bouzidi45, Dr Temi Lampejo45, Dr Malur Sudhanva45, Cassie Breen47, Dr Graciela Sluga48, Dr Shazaad SY Ahmad49,70, Dr Ryan P George49, Dr Nicholas W Machin49,70, Debbie Binns50, Victoria James50, Dr Rachel Blacow55, Dr Lindsay Coupland58, Dr Louise Smith59, Dr Edward Barton60, Debra Padgett60, Garren Scott60, Dr Aidan Cross61, Dr Mariyam Mirfenderesky61, Jane Greenaway62, Kevin Cole64, Phillip Clarke67, Nichola Duckworth67, Sarah Walsh67, Kelly Bicknell68, Robert Impey68, Dr Sarah Wyllie68, Richard Hopes70, Dr Chloe Bishop72, Dr Vicki Chalker72, Dr Ian Harrison72, Laura Gifford74, Dr Zoltan Molnar77, Dr Cressida Auckland79, Dr Cariad Evans85,109, Dr Kate Johnson85,109, Dr David G Partridge85,109, Dr Mohammad Raza85,109, Paul Baker86, Prof Stephen Bonner86, Sarah Essex86, Leanne J Murray86, Andrew I Lawton87, Dr Shirelle Burton-Fanning89, Dr Brendan AI Payne89, Dr Sheila Waugh89, Andrea N Gomes91, Maimuna Kimuli91, Darren R Murray91, Paula Ashfield92, Dr Donald Dobie92, Dr Fiona Ashford93, Dr Angus Best93, Dr Liam Crawford93, Dr Nicola Cumley93, Dr Megan Mayhew93, Dr Oliver Megram93, Dr Jeremy Mirza93, Dr Emma Moles-Garcia93, Dr Benita Percival93, Megan Driscoll96, Leah Ensell96, Dr Helen L Lowe96, Laurentiu Maftei96, Matteo Mondani96, Nicola J Chaloner99, Benjamin J Cogger99, Lisa J Easton99, Hannah Huckson99, Jonathan Lewis99, Sarah Lowdon99, Cassandra S Malone99, Florence Munemo99, Manasa Mutingwende99, Roberto Nicodemi99, Olga Podplomyk99, Thomas Somassa99, Dr Andrew Beggs100, Dr Alex Richter100, Claire Cormie102, Joana Dias102, Sally Forrest102, Dr Ellen E Higginson102, Mailis Maes102, Jamie Young102, Dr Rose K Davidson103, Kathryn A Jackson107, Dr Lance Turtle107, Dr Alexander J Keeley109, Prof Jonathan Ball113, Timothy Byaruhanga113, Dr Joseph G Chappell113, Jayasree Dey113, Jack D Hill113, Emily J Park113, Arezou Fanaie114, Rachel A Hilson114, Geraldine Yaze114 and Stephanie Lo116
Sequencing and analysis:
Safiah Afifi10, Robert Beer10, Joshua Maksimovic10, Kathryn McCluggage10, Karla Spellman10, Catherine Bresner11, William Fuller11, Dr Angela Marchbank11, Trudy Workman11, Dr Ekaterina Shelest13,81, Dr Johnny Debebe18, Dr Fei Sang18, Dr Marina Escalera Zamudio23, Dr Sarah Francois23, Bernardo Gutierrez23, Dr Tetyana I Vasylyeva23, Dr Flavia Flaviani31, Dr Manon Ragonnet-Cronin39, Dr Katherine L Smollett42, Alice Broos53, Daniel Mair53, Jenna Nichols53, Dr Kyriaki Nomikou53, Dr Lily Tong53, Ioulia Tsatsani53, Prof Sarah O'Brien54, Prof Steven Rushton54, Dr Roy Sanderson54, Dr Jon Perkins55, Seb Cotton56, Abbie Gallagher56, Dr Elias Allara70,102, Clare Pearson70,102, Dr David Bibby72, Dr Gavin Dabrera72, Dr Nicholas Ellaby72, Dr Eileen Gallagher72, Dr Jonathan Hubb72, Dr Angie Lackenby72, Dr David Lee72, Nikos Manesis72, Dr Tamyo Mbisa72, Dr Steven Platt72, Katherine A Twohig72, Dr Mari Morgan74, Alp Aydin75, David J Baker75, Dr Ebenezer Foster-Nyarko75, Dr Sophie J Prosolek75, Steven Rudder75, Chris Baxter77, Sílvia F Carvalho77, Dr Deborah Lavin77, Dr Arun Mariappan77, Dr Clara Radulescu77, Dr Aditi Singh77, Miao Tang77, Helen Morcrette79, Nadua Bayzid96, Marius Cotic96, Dr Carlos E Balcazar104, Dr Michael D Gallagher104, Dr Daniel Maloney104, Thomas D Stanton104, Dr Kathleen A Williamson104, Dr Robin Manley105, Michelle L Michelsen105, Dr Christine M Sambles105, Dr David J Studholme105, Joanna Warwick-Dugdale105, Richard Eccles107, Matthew Gemmell107, Dr Richard Gregory107, Dr Margaret Hughes107, Charlotte Nelson107, Dr Lucille Rainbow107, Dr Edith E Vamos107, Hermione J Webster107, Dr Mark Whitehead107, Claudia Wierzbicki107, Dr Adrienn Angyal109, Dr Luke R Green109, Dr Max Whiteley109, Emma Betteridge116, Dr Iraad F Bronner116, Ben W Farr116, Scott Goodwin116, Dr Stefanie V Lensing116, Shane A McCarthy116,102, Dr Michael A Quail116, Diana Rajan116, Dr Nicholas M Redshaw116, Carol Scott116, Lesley Shirley116 and Scott AJ Thurston116
Software and analysis tools:
Dr Will Rowe43, Amy Gaskin74, Dr Thanh Le-Viet75, James Bonfield116, Jennifier Liddle116 and Andrew Whitwham116
1Barking, Havering and Redbridge University Hospitals NHS Trust, 2Barts Health NHS Trust, 3Belfast Health & Social Care Trust, 4Betsi Cadwaladr University Health Board, 5Big Data Institute, Nuffield Department of Medicine, University of Oxford, 6Blackpool Teaching Hospitals NHS Foundation Trust, 7Bournemouth University, 8Cambridge Stem Cell Institute, University of Cambridge, 9Cambridge University Hospitals NHS Foundation Trust, 10Cardiff and Vale University Health Board, 11Cardiff University, 12Centre for Clinical Infection and Diagnostics Research, Department of Infectious Diseases, Guy's and St Thomas' NHS Foundation Trust, 13Centre for Enzyme Innovation, University of Portsmouth, 14Centre for Genomic Pathogen Surveillance, University of Oxford, 15Clinical Microbiology Department, Queens Medical Centre, Nottingham University Hospitals NHS Trust, 16Clinical Microbiology, University Hospitals of Leicester NHS Trust, 17County Durham and Darlington NHS Foundation Trust, 18Deep Seq, School of Life Sciences, Queens Medical Centre, University of Nottingham, 19Department of Infectious Diseases and Microbiology, Cambridge University Hospitals NHS Foundation Trust, 20Department of Medicine, University of Cambridge, 21Department of Microbiology, Kettering General Hospital, 22Department of Microbiology, South West London Pathology, 23Department of Zoology, University of Oxford, 24Division of Virology, Department of Pathology, University of Cambridge, 25East Kent Hospitals University NHS Foundation Trust, 26East Suffolk and North Essex NHS Foundation Trust, 27East Sussex Healthcare NHS Trust, 28Gateshead Health NHS Foundation Trust, 29Great Ormond Street Hospital for Children NHS Foundation Trust, 30Great Ormond Street Institute of Child Health (GOS ICH), University College London (UCL), 31Guy's and St. Thomas’ Biomedical Research Centre, 32Guy's and St. Thomas’ NHS Foundation Trust, 33Hampshire Hospitals NHS Foundation Trust, 34Health Services Laboratories, 35Heartlands Hospital, Birmingham, 36Hub for Biotechnology in the Built Environment, Northumbria University, 37Hull University Teaching Hospitals NHS Trust, 38Imperial College Healthcare NHS Trust, 39Imperial College London, 40Infection Care Group, St George’s University Hospitals NHS Foundation Trust, 41Institute for Infection and Immunity, St George’s University of London, 42Institute of Biodiversity, Animal Health & Comparative Medicine, 43Institute of Microbiology and Infection, University of Birmingham, 44Isle of Wight NHS Trust, 45King's College Hospital NHS Foundation Trust, 46King's College London, 47Liverpool Clinical Laboratories, 48Maidstone and Tunbridge Wells NHS Trust, 49Manchester University NHS Foundation Trust, 50Microbiology Department, Buckinghamshire Healthcare NHS Trust, 51Microbiology, Royal Oldham Hospital, 52MRC Biostatistics Unit, University of Cambridge, 53MRC-University of Glasgow Centre for Virus Research, 54Newcastle University, 55NHS Greater Glasgow and Clyde, 56NHS Lothian, 57NIHR Health Protection Research Unit in HCAI and AMR, Imperial College London, 58Norfolk and Norwich University Hospitals NHS Foundation Trust, 59Norfolk County Council, 60North Cumbria Integrated Care NHS Foundation Trust, 61North Middlesex University Hospital NHS Trust, 62North Tees and Hartlepool NHS Foundation Trust, 63North West London Pathology, 64Northumbria Healthcare NHS Foundation Trust, 65Northumbria University, 66NU-OMICS, Northumbria University, 67Path Links, Northern Lincolnshire and Goole NHS Foundation Trust, 68Portsmouth Hospitals University NHS Trust, 69Public Health Agency, Northern Ireland, 70Public Health England, 71Public Health England, Cambridge, 72Public Health England, Colindale, 73Public Health Scotland, 74Public Health Wales, 75Quadram Institute Bioscience, 76Queen Elizabeth Hospital, Birmingham, 77Queen's University Belfast, 78Royal Brompton and Harefield Hospitals, 79Royal Devon and Exeter NHS Foundation Trust, 80Royal Free London NHS Foundation Trust, 81School of Biological Sciences, University of Portsmouth, 82School of Health Sciences, University of Southampton, 83School of Medicine, University of Southampton, 84School of Pharmacy & Biomedical Sciences, University of Portsmouth, 85Sheffield Teaching Hospitals NHS Foundation Trust, 86South Tees Hospitals NHS Foundation Trust, 87Southwest Pathology Services, 88Swansea University, 89The Newcastle upon Tyne Hospitals NHS Foundation Trust, 90The Queen Elizabeth Hospital King's Lynn NHS Foundation Trust, 91The Royal Marsden NHS Foundation Trust, 92The Royal Wolverhampton NHS Trust, 93Turnkey Laboratory, University of Birmingham, 94 University College London Division of Infection and Immunity, 95University College London Hospital Advanced Pathogen Diagnostics Unit, 96University College London Hospitals NHS Foundation Trust, 97University Hospital Southampton NHS Foundation Trust, 98University Hospitals Dorset NHS Foundation Trust, 99University Hospitals Sussex NHS Foundation Trust, 100University of Birmingham, 101University of Brighton, 102University of Cambridge, 103University of East Anglia, 104University of Edinburgh, 105University of Exeter, 106University of Kent, 107University of Liverpool, 108University of Oxford, 109University of Sheffield, 110University of Southampton, 111University of St Andrews, 112Viapath, Guy's and St Thomas' NHS Foundation Trust, and King's College Hospital NHS Foundation Trust, 113Virology, School of Life Sciences, Queens Medical Centre, University of Nottingham, 114Watford General Hospital, 115Wellcome Centre for Human Genetics, Nuffield Department of Medicine, University of Oxford, 116Wellcome Sanger Institute, 117West of Scotland Specialist Virology Centre, NHS Greater Glasgow and Clyde, 118Whittington Health NHS Trust.
Funding
The study was funded by the Department of Health and Social Care in England. Sequencing was provided through the COVID-19 Genomics UK Consortium (COG-UK) which is supported by funding from the Medical Research Council (MRC) part of UK Research & Innovation (UKRI), the National Institute of Health Research (NIHR) [grant code: MC_PC_19027], and Genome Research Limited, operating as the Wellcome Sanger Institute.
Author information
Authors and Affiliations
Consortia
Contributions
PE and CAD are joint corresponding authors. OE, SR, MC-H, CAD and PE conceived the study and the analytical plan. OE and LdOM performed the statistical analyses. OE, HWang, DH, BB and JJ curated the data. CA, DA, WB, GT, GC, HW, AD provided insights into the study design and results interpretation. AJP generated the sequencing data. AD and PE obtained funding. All authors revised the manuscript for important intellectual content and approved the submission of the manuscript. PE had full access to the data and takes responsibility for the integrity of the data and the accuracy of the data analysis and for the decision to submit for publication. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
We obtained research ethics approval from the South Central-Berkshire B Research Ethics Committee (IRAS ID: 283787). All methods were carried out in accordance with relevant guidelines and regulations. Informed consent was obtained from all participants or their parent/guardian for minors. During initial registration for the study participants are asked “Are you willing to take part in this study?/Are you willing for your child to take part in this study?” with possible answers being “1. Yes, I want to take part in this study” or “2. No, I do not want to take part.”. Those who answered “2. No, I do not want to take part.” were not sent testing kits and did not participate further in the study. Full registration forms for all rounds of REACT-1 are available here: https://www.imperial.ac.uk/medicine/research-and-impact/groups/react-study/for-researchers/react-1-study-materials/.
Consent for publication
Not applicable.
Competing interests
We declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
REACT-1 sequence accession numbers for GISAID and the European Nucleotide Archive.
Additional file 2: Table S1.
Lineages detected in rounds 14 and 15 of REACT-1. Table S2. Estimates of Shannon diversity for England, and by region for rounds 14 and 15 of REACT-1. Table S3. Regional distribution of AY.4 (round 14 and round 15), AY.4.2 (round 15) and B.1.617.2 (round 15). Table S4. Raw numbers of all lineages by region for round 14 and 15 of REACT-1. Table S6. Estimated P-value for the presence of clustering for all lineages with more than a single sample in an individual round, for round 14 and 15 of REACT-1. Table S6. Distribution of AY.4 (round 14 and round 15), AY.4.2 (round 15) and B.1.617.2 (round 15) by age group. Table S7. Raw numbers of all lineages by age group for round 14 and 15 of REACT-1. Table S8. Estimated growth rate in the log odds of every lineage detected relative to all other lineages from round 14 to 15 of REACT-1. Table S9. Mean N- and E-gene Ct value for the eight most prevalent lineages as inferred from Gaussian regression. Table S10. Symptom status by lineage for the eight most prevalent lineages in rounds 14 and 15 of REACT-1. Table S11. Multivariable logistic regression models to determine the effect of the lineage AY.4.2 on the odds of an individual reporting any of the most predictive COVID-19 symptoms relative to AY.4. Table S12. Multivariable gaussian regression model to determine the effect of lineage and round of the study on mean mutation rate. Table S13. Average inter-region migration rate, inferred from a mugration model run on a time-resolved phylogenetic tree, for the periods of rounds 14 and 15, round 14 and round 15
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Eales, O., Page, A.J., de Oliveira Martins, L. et al. SARS-CoV-2 lineage dynamics in England from September to November 2021: high diversity of Delta sub-lineages and increased transmissibility of AY.4.2. BMC Infect Dis 22, 647 (2022). https://doi.org/10.1186/s12879-022-07628-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-022-07628-4