
Abstract
Background
Loop isothermal amplification (LAMP) has recently been proposed as a point-of-care diagnostic tool to detect acute infectious pathogens; however, this technique embeds risk of generating false-positive results. Whereas, with abilities to accurately recognize specific sequence, the CRISPR/Cas12a can forms complexes with cognate RNA sensors and cleave pathogen’s DNA targets complimerntary to its cognate RNA, afterward acquiring the collateral activity to unbiasedly cut nearby off-target fragments. Therefore, if relevant fluorescent-quencher-nucleic probes are present in the reaction, the non-specific cleavage of probes releases fluorescences and establish diagnostic read-outs.
Methods
The MetA gene of N. meningitidis was selected as target to optimize the LAMP reaction, whereas pseudo-dilution series of N. meningitidis gemonics DNA was used to establish the detection limit of LAMP/Cas12a combination assay. The diagnostic performance of established LAMP/Cas12a combination assay was validated in comparation with standard real-time PCR on 51 CSF samples (14 N. meningitidis confirmed patients and 37 control subjects).
Results
In relevant biochemical conditions, CRISPR-Cas12a and LAMP can work synchronously to accurately identify genetics materials of Nesseria menitigistis at the level 40 copies/reaction less than 2 h.
Conclusions
In properly optimized conditions, the CRISPR-Cas12a system helps to alleviate false positive result hence enhancing the specificity of the LAMP assays.
Similar content being viewed by others
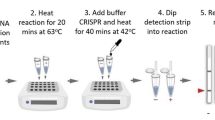

Background
Neisseria meningitidis is a Gram-negative bacterium that causes severe meningitis and sepsis. These diseases require fast and accurate diagnostics to indicate proper antimicrobial therapies [1, 2]. So far, together with blood culture, polymerase chain reaction (PCR) is recommended as a routine technique for the diagnostic confirmation [3, 4]. PCR requires laboratories with sophisticated infrastructures and well-trained personnel, therefore making challenges for deploying in limited-resource areas. Loop-mediated isothermal amplification (LAMP) based approaches have been used to detect pathogens [5]. LAMP-based assays are faster and require no sophisticated instruments or/and skilled personnel, therefore having the advantage to use as on-site diagnostic device [6].
LAMP can achieve PCR’s sensitivity without complicated thermocycling, some LAMP assays can be completed within 30 min. However, LAMP detection step acquires non-specific indicators (such as Mg2+, intercalating dyes, labelled primers) that cannot distinguish spurious amplicons [7,8,9]. We documented several phenomena that real-time PCR protocols [4, 10] could not recapitulate positive results gained by LAMP reactions and some LAMP positive cases lacked meningitidis specific clinical symptoms [1].
We suspected that LAMP assay might embed risks of generating false positive [7,8,9]. Whereas, with abilities to accurately recognize specific sequences, the CRISPR-Cas system holds promising potentials to tackle the above-mentioned problem: In this system, the DNAse cas12a forms a complex with their cognate CRISPR RNAs to induce the cleavage of pathogen’s RNA or DNA in a sequence-specific manner. Afterwards, the collateral transcleavage activity is also triggered to unbiasedly cut the nearby off-target fragments. If relevant fluorescent-quencher-nucleic probes are present in the reaction, the non-target cleavage of probes will release fluorescent signals and establish diagnostics read-outs [11,12,13]. Thus, in this study, we have established an effective method combining the LAMP assay and the CRISPR-Cas12a system for the diagnosis of patients infected with Neisseria meningitidis.
Results
We first performed isothermal amplification assay using Bst DNA Polymerase with primers specific for MetA gene of N. meningitidis. The reaction products were resolved against 1.5% agarose gel. It is impossible to distinguish the electrophoretic banding pattern between human DNA, E. coli DNA or N. meningitidis DNA (Fig. 1 upper-left panel and Additional file 1: Fig. S1). However, once these products were treated with CRISPR-Cas12a with gRNA sequence complementary to MetA gene of N. meningitidis, only fluorescent signals from samples with N. meningitidis DNA was recorded. Thus, treatment of CRISPR-Cas12a helps to alleviate false-positive results by single-use of LAMP assay.

Addition of CRISPR-Cas12a help to alleviate false positive acquired by single use of LAMP performance: (upper left panel) the product mixture of LAMP assay was colometric indicated by addition of 100 uM Hydroxy naphthol blue (HNB Sigma—Singapore) or (downer left panel) resolved against 1.2% agarose gel electrophoresis. Right panel—the same product mixture of LAMP was treated with CRISPR-Cas12a
To evaluate the detection limit of LAMP/CRISPR-Cas12a combination for the detection of N. meningitidis DNA, we spiked series of 0, 40, 400, 4,000, 40,000 and 400,000 copies of N. meningitidis PCR amplicon into 25 mM Tris–EDTA pH 8 containing the background of 108 copies of E. coli PCR amplicon/ul (Additional file 2). These dilution points were used as the templates for Bst DNA Polymerase isothermal amplification at 55 °C for 45 min then treated with CRISPR-Cas12a (1.0 µM Cas12a per reaction), in the presence of 0.25 µM guide RNA and 0.25 µM fluorescence labelled reporter at 55 °C for 30 min; the fluorescent signal was recorded in 510 nm in Roche light cycler 480. The fluorescence was detected at all prepared dilution points even at the lowest level of 40 copies of N. meningitidis (Fig. 2).

Detection limit and diagnostics performance of LAMP/CRISPR-Cas12a for identification of N. meningitidis DNA. Left panel: Detection limit of LAMP/CRISPR-Cas12a for identification of N. meningitidis DNA: the fluorescent signals acquired by Bst DNA Polymerase based isothermally amplifying at 55 °C for 30 min on pseudo-samples of 0, 40, 400, 4000, 40,000 and 400,000 and 400,000 copies of N. menitigitidis PCR amplicon spiked into 25 mM Tris–EDTA pH 8 containing the background of 108 copies E. coli PCR amplicon
To validate the clinical performance, we applied the newly established procedure to identify N. meningitidis from 51 CFS samples from N. meningitidis suspected patients. The standard conventional realtime PCR assay with MetA genes as a molecular target was also used to confirm the presence of N. meningitidis DNA. Realtime PCR identified N. meningitidis DNA from 13 out of 51 recruited CFS samples (Additional file 1: Fig. S3). Whereas, single-use of LAMP assay identified 18 cases positive with N. menitigitidis, in which, five cases did not match to either result acquired by real-time PCR or patients’ clinical symptoms and were considered as false positive. However, when LAMP reaction mixtures from 51 CFS samples were treated with CRISPR-Cas12a, only 13 cases were positive. Importantly, all of these 13 cases were matched to the results gained by conventional PCR (Table 1).
Discussion
Single-use of LAMP embeds high risk of false-positive signals that challenge the employing LAMP-based assays into clinical practices [7, 9]. However, LAMP posscess strong intrinsic amplification potential and it is simple to operate, hence would benefit the communities if the LAMP’s weaknesses are somehow solved. Our data revealed that the sequential treatment LAMP products by CRISPR-Cas12a under the guidance of specific gRNA sequence can abolish non-specific signals. This technical integration of two enzymes, in one side help to sustain the strong amplification potential of LAMP on the other side, significantly enhances the specificity of diagnostic procedures.
Previous study has reported that trehalose is an exceptional protein thermal stability stabilizer [14], and we also believe and can recapitulate the previous findings (Additional file 1: Fig. S2) and found that trehalose helps CRISPR-Cas12a to sustain their activity in a single trehalose containing buffer at 55 °C (Additional file 1: Fig. S2). This condition omits buffer replacement from the LAMP into CRISPR assay thereby reducing sample handling and contamination risk. However, in various tested biochemical environments, CRISPR-Cas12a strongly inhibits isothermal DNA polymerases (Bst and Bsu), therefore, we were not successful in coupling isothermal enzymes and CRISPR-Cas12a into single reaction tubes. Further studies are needed to mitigate the inhibitory effect of CRISPR-Cas12a to Bst or Bsu DNA polymerase, thereby combining CRISPR-Cas12a with isothermal amplification into single tube diagnostic device.
Conclusion
The sequential combination of LAMP and CRISPR-Cas12a can alleviate false-positive acquired by single use of LAMP performance.
Availability of data and materials
Data and supporting materials associated with this study will be shared upon request.
Abbreviations
- LAMP:
-
Loop isothermal amplification
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- PCR:
-
Polychain reaction
References
Rosenstein NE, Perkins BA, Stephens DS, Popovic T, Hughes JM. Meningococcal disease. N Engl J Med. 2001;344(18):1378–88.
Stephens DS, Greenwood B, Brandtzaeg P. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet. 2007;369(9580):2196–210.
Laboratory Manual for the Diagnosis of Meningitis caused by Neisseria meningitidis, Streptococcus pneumoniae and Haemophilus influenzae http://www.who.int/emc.
Taha MK, Alonso JM, Cafferkey M, Caugant DA, Clarke SC, Diggle MA, Fox A, Frosch M, Gray SJ, Guiver M, et al. Interlaboratory comparison of PCR-based identification and genogrouping of Neisseria meningitidis. J Clin Microbiol. 2005;43(1):144–9.
Wong YP, Othman S, Lau YL, Radu S, Chee HY. Loop-mediated isothermal amplification (LAMP): a versatile technique for detection of micro-organisms. J Appl Microbiol. 2018;124(3):626–43.
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28(12):E63.
Schneider L, Blakely H, Tripathi A. Mathematical model to reduce loop mediated isothermal amplification (LAMP) false-positive diagnosis. Electrophoresis. 2019;40(20):2706–17.
Suleman E, Mtshali MS, Lane E. Investigation of false positives associated with loop-mediated isothermal amplification assays for detection of Toxoplasma gondii in archived tissue samples of captive felids. J Vet Diagn Invest. 2016;28(5):536–42.
Hardinge P, Murray JAH. Reduced false positives and improved reporting of loop-mediated isothermal amplification using quenched fluorescent primers. Sci Rep. 2019;9(1):7400.
Diene SM, Bertelli C, Pillonel T, Jacquier N, Croxatto A, Jaton K, Greub G. Comparative genomics of Neisseria meningitidis strains: new targets for molecular diagnostics. Clin Microbiol Infect. 2016;22(6):568 (e561-567).
Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ, Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018;360:439.
Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356(6336):438–42.
Chen JS, Ma E, Harrington LB, Da Costa M, Tian X, Palefsky JM, Doudna JA. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018;360(6387):436–9.
Kaushik JK, Bhat R. Why is trehalose an exceptional protein stabilizer? An analysis of the thermal stability of proteins in the presence of the compatible osmolyte trehalose. J Biol Chem. 2003;278(29):26458–65.
Acknowledgements
Not applicable.
Funding
This study was partially funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under the Grant number 108.06-2017.21 and partial funded by The Vietnamese Ministry of National Defence under the contract number 364/2020/HD-NCKHCN. The funding agencies had no role in the study design, data collection and analysis, decision to publish, and/or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
NTT, LHS, MHB designed and supervised the studies. LHPS and TXH, DTQ conducted the experiments. NTT, LHS, LHPS, TXH analysed the data and wrote the manuscript. LHS, MHB contributed to provide materials. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study and its accosciated method of consent were submitted for regulatory approval to the Institutional Review Board of the 108 Military Central Hospital in Hanoi and were approved. The Ethical Committee of the 108 Military Central Hospital, Hanoi, provided ethical approval for the study. Informed written consent was obtained from all study participants or from their parents/guardians if the study participant was in an unconscious condition.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Study design and supplementary data.
Additional file 2.
Experiment procedure.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Trung, N.T., Son, L.H.P., Hien, T.X. et al. CRISPR-Cas12a combination to alleviate the false-positive in loop-mediated isothermal amplification-based diagnosis of Neisseria meningitidis. BMC Infect Dis 22, 429 (2022). https://doi.org/10.1186/s12879-022-07363-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-022-07363-w