
Abstract
Background
In this study, the association between human papillomavirus (HPV) infection and related cervical intraepithelial neoplasia (CIN) or cervical cancer and vaginal microbiome was evaluated in Chinese cohorts.
Methods
The vaginal bacterial composition of five groups, HPV-infected women without CINs (HPV, n = 78), women with low-grade squamous intraepithelial lesions (LSIL, n = 51), women with high-grade squamous intraepithelial lesions (HSIL, n = 23), women with invasive cervical cancer (Cancer, n = 9) and healthy women without HPV infection (Normal, n = 68), was characterized by deep sequencing of barcoded 16S rRNA gene fragments (V3–4) using Illumina MiSeq.
Results
HPV infection increased vaginal bacterial richness and diversity regardless of the status of CINs. The vaginal bacterial richness and diversity were further augmented in women with cervical cancer. Lactobacillus was the most abundant genus in all groups. HPV infection had a negative influence on the abundances of Lactobacillus, Gardnerella and Atopobium. Accordingly, HPV infection increased the relative abundance of Prevotella, Bacillus, Anaerococcus, Sneathia, Megasphaera, Streptococcus and Anaerococcus. The increased proportions of Bacillus, Anaerococcus and the reduced abundance of Gradnerella vaginalis were probably related with the progression of CINs severity. HPV infection without CINs or cancerous lesions was strongly associated with Megasphaera. The most abundant bacterium in the LSIL group was Prevotella amnii. However, Prevotella timonensis, Shuttleworthia and Streptococcaceae at the family level were three taxa related to HSIL. Furthermore, more taxa were associated with the Cancer group including Bacillus, Sneathia, Acidovorax, Oceanobacillus profundus, Fusobacterium, Veillonellaceae at the family level, Anaerococcus and Porphyromonas uenonis. Samples in the Normal group were mostly assigned to CST III. HPV infection converted the vaginal bacterial community structure from CST III to CST IV. Furthermore, the proportions of CST IV were gradually augmented with the progression of the severity of CINs.
Conclusions
This work interpreted the differential vaginal bacteria under HPV infection and various precancerous or cancerous lesions in a Chinese cohort. We distinguished the specific microbes and the vaginal bacterial structure that were related with the progression of CINs severity in Chinese women.
Similar content being viewed by others

Background
Human papillomavirus (HPV) is regarded as one of the most common sexually transmitted agents in cervical intraepithelial neoplasia (CIN) and cervical cancer [1]. High-risk subtypes of HPV contribute to 99% of cervical neoplasia [1]. However, it is known that high-risk HPV infection is necessary but not sufficient for the development of CINs or cervical cancer [2,3,4]. Many other events, such as multiple sexual partners, early initiation of sexual activity and co-infection with other sexually transmitted infections, have been associated with higher risk of HPV infection in the genital tract [5,6,7].
In the female genital tract, a healthy vaginal status is commonly associated with low microbial diversity and prevalence of Lactobacillus. Lactobacillus prevents colonization of other bacterial pathogens through production of lactic acid, hydrogen peroxide (H2O2) and bacteriocin in the vagina, and therefore keeps integrity of mucosal barriers against virus and opportunistic bacteria [4, 8, 9]. The vaginal microbial profile of women could by classified into five community state types (CSTs) by hierarchical taxonomic clustering, in which CSTs I, II III and V are predominated by L. crispatus, L. iners, L. jensenii and L. gasseri respectively, while CST IV is depleted of Lactobacillus and enriched with anaerobic bacteria like Gardnerella, Megasphera, Sneathia, Prevotella, etc. [10]. Commensal vaginal Lactobacillus species are thought to defend against many pathogens, such as Candida infection [11, 12], sexually transmitted diseases [13], urinary system infections [14, 15] and human immunodeficiency virus (HIV) infection [16]. However, L. iners has many properties different from other Lactobacillus spp., for example unable to produce H2O2, and it often predominates in the presence of HPV infection [4, 17, 18] and CIN [18, 19].
Bacterial vaginosis (BV) is a cluster of microbial disorders characterized by a decrease in Lactobacillus and their replacement by high concentrations of other anaerobic bacteria, with a microbial community structure in accordance with CST IV [9]. BV is associated with a higher risk of miscarriage, preterm premature rupture of membranes and a higher susceptibility to sexually transmitted infections, as HPV infection [20,21,22]. Some studies to date have reported that vaginal microbiome (VM) plays an important role in the persistence of the HPV infection and the subsequent development of cervical precancerous or cancerous lesions [2, 4, 8, 17,18,19, 23,24,25,26,27,28]. Increasing VM diversity is associated with advancing CINs severity and viral persistence [26]. The potential mechanisms could be linked to less production of protective lactic acid, H2O2 and bactriocin by Lactobacillus, disruption of mucosal integrity which may aid viral entry, higher levels of oxidative stress induced by dybiosis [8]. Particular species like Sneathia spp. have a probable pathological role in HPV acquisition and persistence through cellular targets such as expression of immunosuppressive cytokines [29]. Therefore, it is considerable to take vaginal microbiome as a promising marker not only for HPV infection but also for cervical precancerous lesions.
Nevertheless, the vaginal communities could be influenced by many other factors, including ethnicity, personal hygiene, sexual behaviors and hormonal levels [10, 30]. Ethnicity is key to shape vaginal bacterial communities [4, 10, 31]. Caucasian and Asian women display a significantly greater prevalence of Lactobacillus in the vagina compared to Hispanic and Black women [8, 10, 31]. To our knowledge, data with regard to the vaginal bacterial composition of Chinese populations are inadequate [8, 18, 32]. The analysis, which is performed in a large cohort of women living in a different country and with supposed different hygiene habits [10], is helpful to reinforce the underlying associations. Furthermore, there are few studies about the association between VM and HPV infection and related CIN diseases in Chinese cohorts using high throughput sequencing method [8, 18, 32].
Hence, the objective of this research is to study the role of VM on the HPV infection and the progression of CIN diseases in Chinese populations. We try to identify the microbiological markers related with HPV infection and CINs severity in these cohorts.
Methods
Study population and sample collection
We included 229 non-pregnant women, 25–69 years of age, who attended gynecological clinics at the Department of Gynecology, Renji Hospital of Shanghai, Jiao Tong University School of Medicine, between May 2016 and November 2016. Non-pregnant women were included irrespective of their phase in their cycle (except for the menstrual period), parity, personal hygiene, smoking habits. No previous medical histories of CIN diseases or cervical cancer and other serious medical problems, such as hepatitis B/C, diabetes, autoimmune diseases, sexually transmitted diseases (chlamydia, gonorrhea, trichomoniasis, genital herpes), HIV or other malignant tumors, were declared. Participants who had vaginal intercourse or vaginal douching within last 3 days of sampling, abnormal metrorrhagia in the previous weeks, or used probiotics, antibiotics or immunosuppressive drugs in the preceding 14 days were excluded.
HPV genotyping test and ThinPrep cytology test (TCT) were carried out in all enrolled patients, using a commercial HPV genotyping kit as previously described [33]. TCT results were interpreted on the basis of Bethesda System criteria [34]. Women with HPV positive and/or TCT ≥ ASCUS accepted the biopsy under the colposcopy examination by two gynecologists. Based on their histopathology, all participants were assigned into five groups as follow (Fig. S1): HPV-infected women without CINs (HPV, n = 78), women with low-grade squamous intraepithelial lesions (LSIL, n = 51), women with high-grade squamous intraepithelial lesions (HSIL, n = 23), women with invasive cervical cancer (Cancer, n = 9) and healthy women without HPV infection (Normal, n = 68).
Sterile swab samples for 16S rRNA sequencing were taken from the lateral and posterior fornix using a sterile speculum as previously described [33].
Total bacterial genomic DNA extraction and MiSeq sequencing
Primers 338F (ACTCCTACGGGAGGCAGCA) and 806R (GGACTACHVGGGTWTCTAAT) were used to amplify the V3–4 hypervariable fragments of the 16S rRNA gene by PCR as previously described [33]. All sequencing was conducted using the Illumina MiSeq platform at Majorbio Biopharm Technology Company (Shanghai).
Sequence analysis
Sequence reads were quality checked by Trimmomatic [35]. OTUs were generated by QIIME and taxonomies were classified using the Ribosomal Database Project (RDP) classifier script (version 2.2) as previously described [33].
As previously described [33], alpha (Chao and Shannon index) and beta indices (unweighted UniFrac distances in Principal coordinates analysis (PCoA)) were calculated by mothur (version v.1.30.1) [36] and vegan package in R [37]. We compared the differences of alpha and beta estimators between two groups by Student’s t-test and ANOSIM test respectively. Heat maps of relative abundance for different taxa were generated using R. The relative abundances of different taxa at different levels between the five groups were calculated by nonparametric Wilcoxon test. Linear discriminant analysis (LDA) effect size (LEfSe) algorithm [38] was used to characterize the potential microbial markers with specific disease phenotypes. Hierarchical clustering analysis was used to classify different vaginal community state types (CSTs) as previously published [31, 39]. Q-values (p-value adjusted by false discovery rate (FDR)) and p-values < 0.05 were considered significant.
Results
Characteristics of the study population
The average ages of each group (HPV, LSIL, HSIL, Cancer, Normal) were 47.78 ± 9.63, 46.00 ± 10.19, 43.70 ± 10.74, 56.11 ± 9.02 and 43.00 ± 8.69 years, respectively. The proportions of 16/18 HPV subtypes in HPV positive groups were as follow: HPV (13/78), LSIL (10/51), HSIL (6/23), Cancer (4/9) (Table S1). The demographic characteristics of each group are presented in the supplementary data (Table S2).
Sequencing results
After filtering low-quality reads, 6,585,141 assembled clean reads were obtained from 229 samples, with an average number of reads per sample of 28,755.64 ± 5389.65 and a mean read length of 444.90 ± 5.89 bp. For normalization, the reads in each sample were randomly subsampled to the lowest number of 20,098 in sample 318_LCQ (HPV group). After removing singletons (the OTUs contained less than 2 reads), 1878 OTUs were identified, ranging from 10 OTUs in sample 4391 (Normal group) to 782 OTUs in sample 152_GPZ (HPV group) (Table S3). The average number of OTUs in HPV-negative group (Normal group: 46) was lower than that in HPV-positive groups (HSIL group: 116; LSIL group: 145; HPV group: 157). We found more OTUs in the samples of Cancer group (Mean = 256.70 ± 174.78), ranging from 51 to 599 (Table S3).
Vaginal microbiota richness and diversity
At the OTU level, microbial richness and diversity were estimated using Chao and Shannon indices, respectively, as shown in Fig. 1. These two indices revealed that HPV infection increased vaginal bacterial richness and diversity. The means of Chao and Shannon indices were much lower in groups HPV-negative (Normal group: 84.02 ± 73.88 and 0.94 ± 0.95, respectively) than in groups HPV-positive (HPV group: 272.26 ± 191.62 and 1.49 ± 1.01, q < 0.01; LSIL group: 231.84 ± 195.39, q < 0.001 and 1.30 ± 0.93, q = 0.052; HSIL group: 185.74 ± 162.62, q < 0.001 and 1.51 ± 0.98, q = 0.02; Cancer group: 367.76 ± 208.63 and 2.47 ± 0.98, q < 0.001, respectively). The means of Chao and Shannon indices were the highest in group Cancer and were significantly higher than in groups HSIL (Chao: 367.76 ± 208.63 vs. 185.74 ± 162.62, q = 0.02, Shannon: 2.47 ± 0.98 vs. 1.51 ± 0.98, q = 0.02), LSIL (Shannon: 2.47 ± 0.98 vs. 1.30 ± 0.93, q = 0.003) and HPV (Shannon: 2.47 ± 0.98 vs. 1.49 ± 1.01, q = 0.02). However, there were no differences found among groups HPV, LSIL and HSIL.

Vaginal bacterial richness and diversity in five groups. a: Chao index; b: Shannon index; Student’s t-test was used to compare differences between two groups; data are presented as the mean ± SD; ***: q ≤ 0.001; **: 0.001 < q < 0.01; *: q < 0.05
Vaginal bacterial structure and beta-diversity in different groups
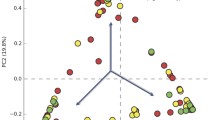
In PCoA, the first two principal components explained 21.18 and 8.83%, respectively, of the variance along the first and second axes, with the Cancer, HSIL, LSIL and HPV samples visually separated from the Normal sample (Fig. 2). Comparison between two groups based on the ANOSIM test revealed that the bacterial structure of groups Cancer, LSIL and HPV were significantly different from that of group Normal (Table S4). Meanwhile, the bacterial structure of groups HSIL and LSIL were also different from that of group HPV (Table S4).

Unweighted UniFrac principal coordinates analysis (PCoA) plot comparing sample distribution for the different groups
Taxonomy of the vaginal microbiota in different groups
Overall, 37 bacterial phyla were recovered across all samples (Fig. S2), and All of the samples were dominated by Firmicutes, Actinobacteria, Bacteroidetes, Fusobacteria and Proteobacteria (Fig. S2). Firmicutes was the most abundant phylum, accounting for 73.15, 68.91, 69.70, 70.43 and 47.47% of the Normal, HPV, LSIL, HSIL and Cancer groups, respectively (Fig. 3a). HPV infection tended to lower the proportion of Firmicutes (Cancer < Normal, q < 0.05; HPV < Normal, q < 0.05). However, there were no differences among other HPV positive groups. Accordingly, the proportions of Bacteroidetes, Fusobacteria and Proteobacteria were significantly increased in the HPV positive groups as compared to the Normal group (Bacacteroidetes: Cancer 15.72% > HSIL 13.1% > HPV 8.28% > Normal 6.94%, Fig. 3b; Fusobacteria: Cancer 15.84% > LSIL 3.04% > HSIL 2.62% > HPV 2.64% > Normal 2.54%, Fig. 3d; Proteobacteria: Cancer 10.39% > HPV 5.36% > LSIL 3.59% > Normal 1.18%, Fig. 3c). There were no differences among the different groups with regard to the proportion of Actinobacteria.

Relative abundance counts of Firmicutes (a), Bacteroidetes (b), Proteobacteria (c) and Fusobacteria (d), which were found to be the most abundant phyla across all samples. The Wilcoxon test was used to compare differences in the abundance of each phylum between two groups. ***: q ≤ 0.001; **: 0.001 < q < 0.01; *: q < 0.05
At the genus level, a total of 749 taxa were found across all samples (Fig. S3), with Lactobacillus being the most dominant genus overall. The proportion of Lactobacillus was reduced in the HPV positive groups (Cancer: 19.72%, q < 0.05; HSIL: 42.99%, q = 0.098; LSIL: 53.71%, q = 0.051; HPV: 49.20%, q < 0.01, respectively) compared to the Normal group (64.93%) (Fig. 4a). However, there were no significant differences among the groups HPV, LSIL, HSIL and Cancer with regard to the relative abundance of Lactobacillus. There were no differences among the different groups with regard to the abundance of Gardnerella (Fig. 4b). In addition, HPV infection had also a negative effect on the abundance of Atopobium, which sharply decreased from 3.12% in the Normal group to 2.07% in the HPV group (q < 0.01), 2.72% in the LSIL group (q < 0.05) (Fig. 4e). Accordingly, HPV infection increased the relative abundance of Prevotella (Normal: 5.91% < HPV: 7.18%, q < 0.05, Fig. 4c), Bacillus (Normal: 0.34% < LSIL: 3.30% < HPV: 5.77% < HSIL: 5.87% < Cancer: 9.18%, q < 0.001, Fig. 4d), Sneathia (Normal: 2.39% < HPV: 2.61% < LSIL: 3.02% < Cancer: 10.03%, q < 0.05, Fig. 4f), Megasphaera (Normal: 1.01% < LSIL: 2.13% < Cancer: 2.30% < HSIL: 2.57% < HPV: 9.18%, q < 0.05, Fig. 4g), Streptococcus (Normal: 1.03% < Cancer: 1.29% < LSIL: 1.35% < HPV: 1.93%, q < 0.05, Fig. 4h) and Anaerococcus (Normal: 1.22% < Cancer: 3.83%, q < 0.05, Fig. 4i). The precancerous lesions increased the proportion of Bacillus, with a greater bacterial abundance in LSIL and HSIL groups than in HPV group (q < 0.05) (Fig. 4d). A higher proportion of Anaerococcus was also found in the group Cancer compared to the precancerous groups (HPV, LSIL and HSIL, q < 0.05) (Fig. 4i). Two taxa, Bacillus and Anaerococcus, were probably related with the progression of CINs severity.

Relative abundance counts of Lactobacillus (a), Gardnerella (b), Prevotella (c), Bacillius (d), Atopobium (e), Sneathia (f), Megasphaera (g), Streptococcus (h) and Anaerococcus (i), which were found to be the most abundant genera across all samples. The Wilcoxon test was used to compare differences in the abundance of each phylum between two groups. ***: q ≤ 0.001; **: 0.001 < q < 0.01; *: q < 0.05
Identification of vaginal microbiological markers in different groups
LEfSe modeling was employed to identify microbiological markers related to HPV infection and CINs severity (Fig. 5). The threshold for the logarithmic LDA model score for discriminative features in this study was 4.0 (p < 0.05). The most abundant genus in the Normal group was Lactobacillus. In addition, other two taxa were also more abundant in the Normal group, Bacilli at the class level and Atopobium vaginae. HPV infection without CIN or cancerous lesions (HPV group) was strongly associated with Megasphaera. The most abundant bacterium in the LSIL group was Prevotella amnii. However, Prevotella timonensis, Shuttleworthia and Streptococcaceae at the family level were three taxa related to HSIL. Furthermore, more taxa were associated with the Cancer group including Bacillus, Sneathia, Acidovorax, Oceanobacillus profundus, Fusobacterium, Veillonellaceae at the family level, Anaerococcus and Porphyromonas uenonis.

The unique taxa and microbiomarkers for different groups. Shown is a histogram of LDA scores computed for features differentially abundant in the five groups
Characteristics of vaginal community state types (CSTs) for different groups
The vaginal bacterial CST analysis visualized by hierarchical clustering revealed that all samples clustered into five major groups: CST I, CST II, CSTII, CST IV and CST V (Fig. 6). The most commonly observed community was CST IV (91/229, 39.74%), followed by CST III (85/229, 37.12%), CST I (44/229, 19.21%), CST II (5/229, 2.18%) and CST V (4/229, 1.75%) as shown in Table 1. The proportions of CSTs in different groups were shown in Table 1. Samples in the Normal group were mostly assigned to CST III (32/68, 47.96%). HPV infection converted the vaginal bacterial community structure from CST III to CST IV, as all of the HPV positive groups were dominated by CST IV (HPV: 32/78, 41.03%; LSIL: 18/51, 35.29%; HSIL: 13/23, 56.52%; Cancer: 8/9, 88.89%, respectively) and presented less CST III (HPV: 28/78, 35.90%; LSIL: 17/51, 33.33%; HSIL: 7/23, 30.43%; Cancer: 1/9, 11.11%, respectively). Furthermore, the proportions of CST IV were gradually augmented with the progression of the severity of CINs (Cancer: 88.89% > HSIL: 56.52% > LSIL: 35.29%).

Heat map of the relative abundance of the 29 most abundant bacterial taxa found in the vaginal bacterial communities of all participants in the study. Ward linkage clustering was used to cluster samples based on their Jensen-Shannon distance. Identified CSTs are labeled as I, II, III, IV and V, according to a previous naming convention [10]
We identified the most abundant species in each sample in Fig. 6, and the distributions of the 29 most predominant species in five groups were documented in Table 2. Seventy-eight samples (34.1%) were predominated by Lactobacillus iners, followed by Lactobacillus crispatus (48 samples, 21%), Gardnerella vaginalis (26 samples, 11.4%), Bacillus unclassified (16 samples, 7%), Sneathia amnii (8 samples, 3.5%) and Prevotella amnii (7 samples, 3.1%). HPV infection was related with the decreased abundance of Lactobacillus iners, Lactobacillus crispatus, Gardnerella vaginalis, Lactobacillus gasseri, Anaerococcus spp., Atopobium unclassified and Porphyromonas somerae as compared to the Normal group (Table 2). In addition, HPV infection was associated with the increased abundance of Bacillus unclassified, Escherichia Shigella, Megasphaera unclassified, Streptococcus unclassified, Lactobacillus jensenii, Sneathia sanguinegens, Bifidobacterium unclassified, Candidatus Mycoplasma, Comamonadaceae unclassified, Veillonella montpellierensis, Faecalibacterium unclassified, Finegoldia unclassified, Fusobacterium mortiferum, Porphyromonas uenonis and Ralstonia pickettii (Table 2). Furthermore, the abundance of Gradnerella vaginalis was gradually reduced with the progression of CINs severity (Cancer 0% < HSIL 4.3% < LSIL 11.8% < HPV 12.8% < Normal 13.2% as shown in Table 2).
Discussion
Our study addressed a not well-elucidated topic about the association between HPV infection and related CINs or cervical cancer and vaginal microbiome in Chinese cohorts. We observed that HPV infection increased vaginal bacterial richness and diversity regardless of the status of CINs. The vaginal bacterial richness and diversity were further augmented in the women with cervical cancer. Lactobacillus was the most abundant genus in all groups. HPV infection had a negative influence on the abundances of Lactobacillus, Gardnerella and Atopobium. Accordingly, HPV infection increased the relative abundance of Prevotella, Bacillus, Anaerococcus, Sneathia, Megasphaera, Streptococcus and Anaerococcus. The increased proportions of Bacillus, Anaerococcus and the reduced abundance of Gradnerella vaginalis were probably related with the progression of CINs severity. HPV infection without CINs or cancerous lesions was strongly associated with Megasphaera. The most abundant bacterium in the LSIL group was Prevotella amnii. However, Prevotella timonensis, Shuttleworthia and Streptococcaceae at the family level were three taxa related to HSIL. Furthermore, more taxa were associated with the Cancer group including Bacillus, Sneathia, Acidovorax, Oceanobacillus profundus, Fusobacterium, Veillonellaceae at the family level, Anaerococcus and Porphyromonas uenonis. Samples in the Normal group were mostly assigned to CST III. HPV infection converted the vaginal bacterial community structure from CST III to CST IV. Furthermore, the proportions of CST IV were gradually augmented with the progression of the severity of CINs.
Most of the studies have proven that HPV infection can increase vaginal bacterial richness and diversity and lower the percentage of Lactobacillus [4, 8, 17, 18, 23, 24, 40, 41], and our results are in agreement with these previous studies. However, a few studies found no difference between HPV positive and negative groups [25, 42]. HPV infection is thought to alter the acidic environment of the vagina, which might promote outbreaks of bacteria [24]. In addition, HPV infection might lead to changes in the vaginal microbiota by inducing host mucosal immune response and genital infalmmation [41, 43]. High genital inflammation with elevated vaginal PH and non-Lactobacillus-dominant VM have been associated with HPV persistence and progression to cervical cancer [44]. But the underlying biological mechanisms are still unclear. On the other hand, Mitra et al. reported that increasing CINs severity was associated with decreasing relative abundance of Lactobacillus and increasing bacterial diversity [26]. Differently, we only observed a higher bacterial richness and diversity in group Cancer than in groups HSIL, LSIL or HPV, but no differences were detected when compared between each two of groups HPV, LSIL and HSIL. Some other studies also found no connection between the diversity of VM and the CINs progression [45, 46]. It is notable that the study of Mitra et al. [26] did not distinguish the influence of HPV infection from precancerous or cancerous lesions on the diversity of VM.
Similarly to the previous studies [2, 4, 17, 18, 23, 24, 40, 42, 47], we found increased abundances of several anaerobic bacteria such as Prevotella, Bacillus, Anaerococcus, Sneathia, Megasphaera, Streptococcus and Anaerococcus in HPV-infected women. We recognized Megasphaera of Firmicutes phylum as the most significant genus related with HPV infection, while Lee et al. identified Sneathia spp. of Fusobacteria phylum as the microbiological marker of HPV infection [24]. Over all, a microbial environment with a higher proportion of anaerobic bacteria and a lower proportion of Lactobaillus spp. is more likely to HPV infection. A surprising finding in this study was that the proportions of Gardnerella and Atopobium, were reduced in HPV-positive women. Gardnerella vaginalis and Atopobium vaginae were thought to be associated with BV [48]. Gao et al. reported that these two taxa were more frequently detected in HPV-infected women [23]. However, the method used in their study was totally different from this research. Another study in a Caucasian cohort proposed Atopobium spp. and sialidase-encoding gene from Gardnerella vaginalis as microbial markers of HPV persistence [40]. It is hypothesized that the enzyme (sialidase) facilitates the destruction of the mucus layer on the vaginal epithelium and entraps anaerobic bacteria such as Prevotella and Atopobium. The potential reason for this different result remains to explore. To distinguish the influence of HPV infection from precancerous or cancerous lesions on vaginal microbiota, we compared the vaginal bacterial composition between group HPV and groups LSIL, HSIL or Cancer. We found that the proportions of two specific taxa, Bacillus and Anaerococcus, were positively related with the progression of CINs severity. Furthermore, we identified respective taxa for different stages of CIN lesions. Mitra et al. also reported that higher levels of Sneathia sanguinegens, Anaerococcus tetradius and Peptostreptococcus anaerobius were characterized in HSIL compared to LSIL [26]. Corresponding to the impact of HPV infection on the vaginal microbes, we also found that the abundance of Gradnerella vaginalis was gradually reduced with the progression of CINs severity. However, some other studies [19, 27] thought that an enrichment of Gradnerella vaginalis and Atopobium vaginae had a higher CIN risk.
In accordance with the results from an Asian population in the study by Ravel et al. [10], the most abundant CST in Normal group was CST III. The most dominant CST in the HPV positive groups (HPV, LSIL, HSIL and Cancer) was CST IV. We observed HPV infection to be associated with an increased proportion of CST IV, and furthermore its proportion was gradually augmented with the progression of the severity of CINs. It has also been reported by two longitudinal studies that the majority of HPV-positive samples were composed of CST IV (dominated by anaerobic bacteria), and CST IV was related with an increased risk of transitioning to an HPV-positive state [24, 49]. Mitra et al. also found that the rate of CST IV was increased 2 fold in women with LSIL, 3 fold in women with HSIL and 4 fold in women with invasive cancer [26]. CST IV is associated with higher levels of amine production, resulting in carcinogens nitrosamine production [50].
The strength of this study is that it interpreted the vaginal microbial compositions of a large cohort of Chinese women with different stages of HPV-related diseases using high throughput sequencing method, which has not yet been well elucidated. We found that HPV infection increased vaginal bacterial richness and diversity regardless of the status of CINs. The specific microbes and the vaginal bacterial structure were related with the progression of CINs severity in Chinese women. The limitations of this study were that it was a cross-sectional study. Hence, we could not conclude any causal relationship between the VM and HPV infection or CIN diseases. We have to conduct longitudinal studies to study relationships between the dynamics of the VM and the persistence or clearance of HPV infection, and the progression or remission of CIN diseases. In addition, the underlying biological mechanisms also need to be detailed.
Conclusion
This work interpreted the differential vaginal bacteria under HPV infection and various precancerous or cancerous lesions in a Chinese cohort. We distinguished the specific microbes and the vaginal bacterial structure that were related with the progression of CINs severity in Chinese women.
Availability of data and materials
All reads obtained were submitted to NCBI Sequence Read Archive (SRA) under the accession number SRP122481 (https://www.ncbi.nlm.nih.gov//bioproject/PRJNA415526).
Abbreviations
- HPV:
-
Human papillomavirus
- LSIL:
-
Low-grade squamous intraepithelial lesion
- HSIL:
-
High-grade squamous intraepithelial lesions
- BV:
-
Bacterial vaginosis
- VM:
-
Vaginal microbiome
- HIV:
-
Human immunodeficiency virus
- ANOSIM:
-
Analysis of similarities
- CST:
-
Community state type
- CIN:
-
Cervical intraepithelial neoplasia
- TCT:
-
ThinPrep cytology test
- OTUs:
-
Operational taxonomic units
- RDP:
-
Ribosomal database project
- PCoA:
-
Principal coordinates analysis
- LEfSe:
-
Linear discriminant analysis (LDA) effect size
- FDR:
-
False discovery rate
References
Muñoz N, Bosch FX, De SS, Herrero R, Castellsagué X, Shah KV, et al. Epidemiologic classification of human papillomavirus types associated with cervical Cancer. N Engl J Med. 2003;348(6):518–27.
Kyrgiou M, Mitra A, Moscicki AB. Does the vaginal microbiota play a role in the development of cervical cancer? Transl Res. 2017;179:168–82.
Champer M, Wong AM, Champer J, Brito IL, Messer PW, Hou JY, et al. The role of the vaginal microbiome in gynaecological cancer. BJOG. 2018;125:309–15.
Curty G, de Carvalho PS, Soares MA. The role of the Cervicovaginal microbiome on the genesis and as a biomarker of premalignant cervical intraepithelial neoplasia and invasive cervical Cancer. Int J Mol Sci. 2019;21(1):222.
Erickson BK, Alvarez RD, Huh WK. Human papillomavirus: what every provider should know. Am J Obstetr Gynecol. 2013;208(3):169–75.
Hariri S, Unger ER, Sternberg M, Dunne EF, Swan D, Patel S, et al. Prevalence of genital human papillomavirus among females in the United States, the National Health and Nutrition Examination Survey, 2003-2006. J Infect Dis. 2011;204(4):566–73.
Hernández-Girón C, Smith JS, Lorincz A, Lazcano E, Hernández-Avila M, Salmerón J. High-risk human papillomavirus detection and related risk factors among pregnant and nonpregnant women in Mexico. Sex Transm Dis. 2005;32(32):613–8.
Mitra A, Macintyre DA, Marchesi JR, Lee YS, Bennett PR, Kyrgiou M. The vaginal microbiota, human papillomavirus infection and cervical intraepithelial neoplasia: what do we know and where are we going next? Microbiome. 2016;4(1):58.
Witkin SS, Linhares IM. Why do lactobacilli dominate the human vaginal microbiota? BJOG. 2016. https://doi.org/10.1111/1471-0528.14390.
Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108:4680–7.
Deidda F, Amoruso A, Nicola S, Graziano T, Pane M, Allesina S, et al. The in vitro effectiveness of Lactobacillus fermentum against different Candida species compared with broadly used azoles. J Clin Gastroenterol. 2016;50:S171.
Prieto D, Pla J. Distinct stages during colonization of the mouse gastrointestinal tract by Candida albicans. Front Microbiol. 2015;6(1):792.
Borgdorff H, Tsivtsivadze E, Verhelst R, Marzorati M, Jurriaans S, Ndayisaba GF, et al. Lactobacillus-dominated cervicovaginal microbiota associated with reduced HIV/STI prevalence and genital HIV viral load in African women. ISME J. 2014;8(9):1781–93.
Kirjavainen PV, Pautler S, Baroja ML, Anukam K, Crowley K, Carter K, et al. Abnormal immunological profile and vaginal microbiota in women prone to urinary tract infections. Clin Vaccine Immunol. 2009;16(1):29–36.
Messano GA. Inadequate antibiotic therapy of genitourinary tract infections could be responsible for viral sexually transmitted diseases. Ann Ig. 2013;25(6):553–4.
Chehoud C, Stieh DJ, Bailey AG, Laughlin AL, Allen SA, McCotter KL, et al. Associations of the vaginal microbiota with HIV infection, bacterial vaginosis, and demographic factors. Aids. 2017;31(7):895–904.
Brotman RM, Shardell MD, Gajer P, Tracy JK, Zenilman JM, Ravel J, et al. Interplay between the temporal dynamics of the vaginal microbiota and human papillomavirus detection. J Infect Dis. 2014;210(11):1723.
Norenhag J, Du J, Olovsson M, Verstraelen H, Engstrand L, Brusselaers N. The vaginal microbiota, human papillomavirus and cervical dysplasia: a systematic review and network meta-analysis. BJOG. 2019. https://doi.org/10.1111/1471-0528.15854.
Oh HY, B-S K, S-S S, J-S K, J-K L, S-Y P, et al. The association of uterine cervical microbiota with an increased risk for cervical intraepithelial neoplasia in Korea. Clin Microbiol Infect. 2015;21(7):674.e1-.e9.
Baldwin EA, Walther-Antonio M, Maclean AM, Gohl DM, Chia N. Persistent microbial dysbiosis in preterm premature rupture of membranes from onset until delivery. PeerJ. 2015;3(1):e1398.
Gillet E, Meys JF, Verstraelen H, Bosire C, De Sutter P, Temmerman M, et al. Bacterial vaginosis is associated with uterine cervical human papillomavirus infection: a meta-analysis. BMC Infect Dis. 2011;11(1):10.
Smith JS, Herrero R, Bosetti C, Munoz N, Bosch FX, Eluf-Neto J, et al. Herpes simplex virus-2 as a human papillomavirus cofactor in the etiology of invasive cervical cancer. J Natl Cancer Inst. 2002;94:1604–13.
Gao W, Weng J, Gao Y, Chen X. Comparison of the vaginal microbiota diversity of women with and without human papillomavirus infection: a cross-sectional study. BMC Infect Dis. 2013;13(1):271.
Lee JE, Lee S, Lee H, Song YM, Lee K, Han MJ, et al. Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort. PLoS One. 2013;8(5):e63514.
Tuominen H, Rautava S, Syrjänen S, Collado MC, Rautava J. HPV infection and bacterial microbiota in the placenta, uterine cervix and oral mucosa. Sci Rep. 2018;8:9787.
Mitra A, Macintyre DA, Lee YS, Smith A, Marchesi JR, Lehne B, et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci Rep. 2015;5:16865.
Godoy-Vitorino F, Romaguera J, Zhao C, Vargas-Robles D, Ortiz-Morales G, Vázquez-Sánchez F, et al. Cervicovaginal Fungi and Bacteria associated with cervical intraepithelial neoplasia and high-risk human papillomavirus infections in a Hispanic population. Front Microbiol. 2018;9:2533. https://doi.org/10.3389/fmicb.2018.02533.
Siqueira JD, Curty G, Xutao D, Hofer CB, Soares EA. Composite analysis of the virome and bacteriome of HIV/HPV co-infected women reveals proxies for immunodeficiency. Viruses. 2019;11(5):422.
Audirac-Chalifour A, Torres-Poveda K, Bahena-Román M, Téllez-Sosa J, Martínez-Barnetche J, Cortina-Ceballos, et al. Cervical microbiome and cytokine profile at various stages of cervical cancer: a pilot study. Plos One. 2016;11(4):e0153274.
Hickey RJ, Zhou X, Settles ML, Erb J, Malone K, Hansmann MA, et al. Vaginal microbiota of adolescent girls prior to the onset of menarche resemble those of reproductive-age women. mBio. 2015;6(2):e00097–15.
Onywera H, Williamson AL, Mbulawa ZZA, Coetzee D, Meiring TL. Factors associated with the composition and diversity of the cervical microbiota of reproductive-age black south african women: a retrospective cross-sectional study. PeerJ. 2019;7(6):e7488.
Wang H, Ma Y, Li R, Chen X, Wan L and Zhao W. Associations of Cervicovaginal Lactobacilli With High-Risk Human Papillomavirus Infection, Cervical Intraepithelial Neoplasia, and Cancer: A Systematic Review and Meta-Analysis. J Infect Dis. 2019;220(8):1243–54.
Chen Y, Hong Z, Wang W, Gu L, Gao H, Qiu L, et al. Association between the vaginal microbe and high-risk human papillomavirus infection in pregnant Chinese women. BMC Infect Dis. 2019;19:677.
Solomon D, Davey D, Kurman R. Bethesda system\terminology for reporting results of cervical cytology. JAMA. 2002;287(287):2114–9.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ.Microbiol. 2009;75(23):7537–41.
R. Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; R. Core Team; 2018. Available online: http://www.R-project.org/. Accessed 1 Nov2018.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. https://doi.org/10.1186/gb-2011-12-6-r60.
Gajer P, Brotman RM, Bai GY, Sakamoto J, Schuette UME, Zhong X, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med. 2012;4(132):132ra52. https://doi.org/10.1126/scitranslmed.3003605.
Paola MD, Sani C, Clemente AM, Iossa A, Perissi E, Castronovo G, et al. Characterization of cervico-vaginal microbiota in women developing persistent high-risk human papillomavirus infection. Sci Rep. 2017;7(1):10200.
Shannon B, Yi TJ, Perusini S, Gajer P, Ma B, Humphrys MS, et al. Association of hpv infection and clearance with cervicovaginal immunology and the vaginal microbiota. Mucosal Immunol. 2017;10(5):1310–9.
Chao XP, Sun TT, Wang S, Fan QB, Shi HH, Zhu L, et al. Correlation between the diversity of vaginal microbiota and the risk of high-risk human papillomavirus infection. Int J Gynecol Cancer. 2019;29(1):28–34.
Woodworth CD. HPV innate immunity. Front Biosci. 2002;7(7):d2058–71.
Łaniewski P, Cui H, Roe DJ, Barnes D, Herbst-Kralovetz MM. Features of the cervicovaginal microenvironment drive cancer biomarker signatures in patients across cervical carcinogenesis. Sci Rep. 2019;9:7333.
Huang X, Li C, Li F, Zhao J, Wan X, Wang K. Cervicovaginal microbiota composition correlates with the acquisition of high-risk human papillomavirus types. Int J Cancer. 2018;143(3):621–34.
Piyathilake CJ, Ollberding NJ, Kumar R, Macaluso M, Alvarez RD, Morrow CD. Cervical microbiota associated with risk of higher grade cervical intraepithelial neoplasia in women infected with high-risk human papillomaviruses. Cancer Prev Res. 2016;9(5):357–66.
Łaniewski P, Barnes D, Goulder A, Cui H, Roe DJ, Chase DM, et al. Linking cervicovaginal immune signatures, HPV and microbiota composition in cervical carcinogenesis in non-Hispanic and Hispanic women. Sci Rep. 2018;8(1):7593.
Menard JP, Fenollar F, Henry M, Bretelle F, Raoult D. Molecular quantification of Gardnerella vaginalis and Atopobium vaginae loads to predict bacterial vaginosis. Clin Infect Dis. 2008;47:33–43.
Jalil EM, Bastos FI, Melli PP, Duarte G, Simoes RT, Yamamoto AY, et al. HPV clearance in postpartum period of HIV-positive and negative women: a prospective follow-up study. BMC Infect Dis. 2013;13(1):1–9.
Bartsch H, Montesano R. Relevance of nitrosamines to human cancer. Carcinogenesis. 1984;5:1381–93.
Acknowledgements
None
Funding
This work was supported by National Key R&D Program of China (2016YFC1302900), National Natural Science Foundation of China (81874101), Shanghai Municipal Education Commission Gaofeng Clinical Medicine Grant Support (20161412), Shanghai Municipal Commission of Health and Family Planning (2017ZZ02016), Shanghai Municipal Commission of Health and Family Planning Program (15GWZK0701) and Science and Technology Commission of Shanghai Municipality (18441904800). The funders did not have any other role in the design of the study, collection, analysis, and interpretation of data and in writing of this manuscript.
Author information
Authors and Affiliations
Contributions
YC, XQ, WD and LQ contributed to the conception and design of the study. YC, XQ, WW, DL, AW, ZH and LG organized the database. YC, XQ and WW performed the data analysis. YC and XQ wrote the first draft of manuscript. YC and XQ contributed equally to this work. All authors contributed to manuscript revision and read and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of Renji Hospital, School of Medicine, Shanghai Jiao Tong University (registration number: 2018–026), and all experiments were performed in accordance with relevant guidelines and regulations. All participants provided written informed consent. Upon enrollment, some of the participants were asked to complete a questionnaire detailing sexual and reproductive health history and hygiene practices with demographic information.
Consent for publication
Not applicable.
Competing interests
We declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Table 1.
HPV genotyping and distribution in five groups. Supplementary Table 2. Characteristics of study population. Supplementary Table 3 Sequences information. Supplementary Table 4. The comparison between two groups tested by ANOSIM test.
Additional file 2: Supplementary figure 1.
Flow chart of 229 participants. Supplementary figure 2: Heat map of relative abundance for the 30 most abundant bacterial phyla found in the vaginal bacterial communities of 5 groups. Supplementary figure 3. Heat map of relative abundance for the 30 most abundant bacterial genus found in the vaginal bacterial communities of 5 groups.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, Y., Qiu, X., Wang, W. et al. Human papillomavirus infection and cervical intraepithelial neoplasia progression are associated with increased vaginal microbiome diversity in a Chinese cohort. BMC Infect Dis 20, 629 (2020). https://doi.org/10.1186/s12879-020-05324-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-020-05324-9