
Abstract
Background
Human influenza represents a major public health concern, especially in south-east Asia where the risk of emergence and spread of novel influenza viruses is particularly high. The BaliMEI study aims to conduct a five year active surveillance and characterisation of influenza viruses in Bali using an extensive network of participating healthcare facilities.
Methods
Samples were collected during routine diagnostic treatment in healthcare facilities. In addition to standard clinical and molecular methods for influenza typing, next generation sequencing and subsequent de novo genome assembly were performed to investigate the phylogeny of the collected patient samples.
Results
The samples collected are characteristic of the seasonally circulating influenza viruses with indications of phylogenetic links to other samples characterised in neighbouring countries during the same time period.
Conclusions
There were some strong phylogenetic links with sequences from samples collected in geographically proximal regions, with some of the samples from the same time-period resulting to small clusters at the tree-end points. However this work, which is the first of its kind completely performed within Indonesia, supports the view that the circulating seasonal influenza in Bali reflects the strains circulating in geographically neighbouring areas as would be expected to occur within a busy regional transit centre.
Similar content being viewed by others

Background
Virus gene sequencing and phylogenetics can be used to study the epidemiological dynamics of rapidly evolving viruses. With complete genome data, it becomes possible to identify and trace individual transmission chains of circulating viruses such as influenza virus within a defined spatiotemporal context. Next generation sequencing (NGS) has been employed in the high-throughput production of complete viral genome data and offers significant opportunities for increasing our understanding of influenza distribution and transmission. To date, NGS has been used on influenza samples as the basis for identification [1,2,3,4] and comparison [5, 6] of full influenza genomic data as well as for applications such as profiling quasi-species and lineage [7, 8] and characterising interactions with the host-cell. [9, 10] The Molecular Epidemiology of Influenza A in Bali project (“BaliMEI”) [11] aims to conduct five years of active surveillance and characterisation of influenza viruses in Bali (2010–2015). The project utilises a network of 21 health facilities across all nine regencies of Bali to collect nasopharyngeal swabs from patients presenting with influenza-like illness. Here we report the whole genome sequencing results and phylogenetic analysis of the first 95 influenza samples from the BaliMEI project.
Indonesia is of key strategic importance for influenza surveillance and research, as it continues to report highly pathogenic avian influenza (H5N1) outbreaks in poultry, along with sporadic cases in humans. [12] However, research on the ecology and evolution of influenza viruses in Indonesia has been severely limited. Within Indonesia, the island province of Bali might be a particular hotspot for the mixing of influenza viruses from different geographic regions and host species and potential genomic reassortment thereof, due to high densities and close proximity of humans, poultry and pigs, along with its status as a popular tourist destination with continuous, high numbers of a transient population. [13, 14] Importantly the preliminary results from the BaliMEI study allow us to assess the degree of agreement between epidemiologically and genetically inferred information, understand the extent of mutation observed between genetically clustered cases and to improve estimates of the extent of diversity within the circulating, clinically presented influenza.
Methods
Sample collection
The samples were taken during routine diagnostic treatment by hospital physicians between July 2010 and July 2013. All laboratory-confirmed (PCR-positive) samples containing influenza virus from 95 patients were collected through the BaliMEI project protocol under ethics approval number: 41/H2.F10/PPM.00/2010 (University of Indonesia) and 441/Skrt/VI/2010 (Udayana University). These included samples submitted from 21 sentinel health facilities (10 government hospitals and 11 urban health centres) across 8 regencies and one provincial capital city of Denpasar. The 64 samples were obtained from the patients concurrently with epidemiological information through a questionnaire containing sample dates, admission and discharge dates, age, sex, timing of hospital admission and discharge. Patient identifiers were removed prior to the transfer of information to a dedicated, secure Data repository. Laboratory specimen numbers (identifying unique specimens) were retained as these were not interpretable outside the laboratory environment.
Sample sequencing
RNA was extracted from collected specimens that had been stored at -70 °C temp using the QIAamp Viral RNA Mini Kit. RNA extracts were amplified using a modified eight-segment method [5, 15] and library prepared using the Illumina library preparation Kit Nexterra XT at the biomolecular laboratory at Udayana University. The sequencing used the Ilumina MiSeq platform at Pandu Biosains Laboratory. The average read depth and average genome coverage were recorded across all segments.
Sequence de novo assembly
Genome assembly and construction of consensus sequences was performed at Indonesia using the Infection response through virus genomics (ICONIC) bioinformatics pipeline for de novo viral sequence assembly [16] developed at University College London (UCL) with phylogenetic analyses to infer transmission performed at the Farr institute of Health Informatics Research. In short, NGS data were subjected to quality control using Trimmomatic 0.33 to remove any primer sequences and trim reads, then reads mapped with SMALT version 0.7.6 (http://www.sanger.ac.uk/science/tools/smalt-0) to the human genome were removed. Quality controlled and filtered read sets were de novo assembled using IVA version 1.0.0 [17]; SAMtools 1.2 [18] and custom scripts were used to create a consensus genome from the assembled fragments (“contigs”). In particular, these scripts utilise BLAST to find the closest matching sequences to the draft segments and use them as templates on which to map unassembled reads with SMALT.
Phylogenetic analysis
The influenza virus sequences were combined with existing sequences retrieved from the NCBI Influenza Virus Resource [19] that represented the range of genetic diversity worldwide during the same period (Additional file 1). Each genome segment was aligned separately using the MUSCLE aligner [20] provided in MEGA version 6.06. [21] Separate alignments were made for H1N1 and H3N2 sequences. Alignments were then trimmed to coding regions, and sequences covering less than 50% of the coding region were removed. Phylogenetic trees were then inferred under a maximum-likelihood (ML) criterion using RAxML version 7.2.8. [22] Maximum likelihood (ML) trees were estimated for all the eight gene segments using the best-fit general time reversible (GTR) model of nucleotide substitution with a gamma distribution of among-site rate variation (with four rate categories, Γ4) and an SPR branch-swapping search procedure implemented in PhyML [23]. Tree robustness was determined through bootstrap analysis of 1000 sequence pseudoreplicates. Trees were visualized using FigTree version 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Results
Among the 95 PCR-positive, patient samples with influenza A, the mean age was 14.65 ± 15.56 years old (range 0.5–40 yo), 60% were male, and 27.4% were submitted from hospital. There were a total of 7 samples collected in 2010, 36 samples in 2011, 25 samples in 2012, and 27 samples in 2013. Influenza due to influenza A viruses mostly occurred in children age 0–4 yo (35.8%), following in children age 5–14 yo (27.4%) and 15–24 yo (12.6%). A summary of these results is shown in Table 1. From the 95 samples, 68 (71.6%) were subtyped as A/H1N1-pdm09, 26 (27.4%) as A/H3N2, and 1 (1.1%) as seasonal A/H1N1 (Fig. 1a). The highest number of samples were collected from Denpasar (32.6%), followed by Badung and Buleleng (14.5% and 14.5% respectively) (Fig. 1b) correlating to their relative population density. There was no correlation observed between the calendar time of sample collection 108 and relative numbers of samples, as all influenza types analysed in the present paper were collected throughout the 2010–2013 period.

Relative distribution of the influenza A types collected in Bali (2010–2013) shown on the pie chart (left). Relative distribution of the influenza A samples collected in Bali, broken down by geographical origin shown in the bar chart (right)
Of the 95 collected, PCR-positive influenza A samples, 62 were of sufficient concentration (Ct < 30) to be further processed through an NGS platform in an attempt to generate more extensive viral genomes with high read-depth coverage (17 samples A/H3N2, 45 samples A/H1N1-pdm09). As a result of the NGS sequencing and the subsequent de novo assembly, 31 complete influenza A genomes were assembled with all 8 genomic segments, while for the remaining samples, there was an average recovery of 5 out of the genomic 8 segments. There was complete correspondence of the influenza A typing between the PCR- and NGS-based methods across all of the samples. Across the samples in which it was possible to build segments, the average read depth was 2443 and the average genome coverage was 80%. In particular, segment 3 had the lowest average depth, 559 reads, and segment 1 the lowest average coverage with 56%; while segment 7 had the highest depth, 6073 reads, and segment 8 the highest coverage with 97%. The genome coverage was calculated against the reference found for each sample segment.
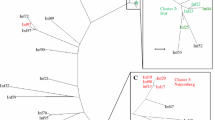
According to the phylogenetic analyses all samples analysed were of the influenza type circulating globally at the time of collection and in particular in proximal countries, such as Singapore, Thailand, Australia and Cambodia. Some samples formed small clusters at the phylogenetic tree end-points indicative of a small-scale localised circulation within Bali, and lack of subsequent transmission to other neighbouring countries or localities (Fig. 2 showing H1N1 samples; all H3N2 samples gave identical distribution – data not shown). However the sample number is small to support any further observations based on the phylogeny alone.

a Maximum-likelihood (ML) tree inferred from the nucleotide sequence of the HA gene for the pandemic H1N1 lineage. The tree is rooted on the A/California/07/2009 strain. Reference isolates for each subclade are shown in grey, with isolates from BaliMEI shown as circles, with colours indicating the year of isolation. Subclades containing BaliMEi isolates are highlighted with a black bar and labelled with the corresponding clade name. The scale bar is given in units of substitution per site. b Magnification of the Balinese cluster within clade 6a, taken from a higher-resolution ML tree containing globally-collected isolates. BaliMEI isolates are labelled in red. The scale bar is in units of substitutions per site. The details of the samples included in this analysis are available in Additional file 1
Discussion
The Influenza patient samples analysed above and collected at Bali, Indonesia, are considered typical of the background seasonal influenza incidence that is ever present in the area. There were no statistically significant differences in the distribution of the samples across the sexes, months of the years or localities. There was a slight tendency towards younger age-groups (0 – 14yo, 46% of samples), where the need for hospitalisation is perhaps more acute than in older age groups, while more densely populated areas and built up areas provided larger numbers of samples in the collection. However a larger sample size would be needed to confirm this observation. The influenza patient samples were typed independently by PCR under routine hospital protocols and by NGS methods; the resulting absolute correlation of the typing is supportive of the NGS-based results. The sequence depth from the influenza samples is adequate for the de novo genomic assembly; however the depth variability across the fragments does not allow the identification of minority variants in a genome-wide approach. The variable depth of the obtained sequences is most likely a reflection of the collection approach where PCR-positive samples were collected during routine diagnostic treatment without a preselection on the Ct value of the viral sample. Quantitative sample pre-selection might result in better NGS output with regards to sequence read-depth but also introduce potential sample bias in the collection. There were no complex reassortant viruses nor any oseltamivir-resistance markers amongst the 62 NGS samples analysed.
The phylogenetic analysis supports the view of these samples being representative of seasonal, circulating Influenza amongst the population in Bali. There were, as expected, strong phylogenetic links with sequences from samples collected in geographically proximal regions, with some of the samples from the same time-period resulting to small clusters at the tree-end points. However more samples would need to be analysed before further claims can be made regarding the influenza transmission in Indonesia from either a local or international perspective. The current paper represents the first ever attempt to utilise NGS technology to characterise clinically relevant samples from Bali, Indonesia as part of the Bali-MEI project. Importantly the current first report reflects work that has been almost entirely taken place within the country as testament to current capacity building efforts and future prospects for even further and more detailed influenza surveillance within Indonesia.
Conclusion
We describe the preliminary results from a five year project on the surveillance and characterization of seasonal influenza samples from healthcare facilities in Bali, Indonesia. This work is the first of its kind completely performed within Indonesia, and can be used as the blueprint for future national molecular surveillance projects. The results support the view that the circulating seasonal influenza in Bali reflects the strains circulating in geographically neighbouring areas as would be expected to occur within a busy regional transit centre.
Abbreviations
- NGS:
-
Next Generation Sequencing
- BaliMEI:
-
Molecular Epidemiology of Influenza in Bali, Indonesia
- PCR:
-
Polymerase Chain Reaction
- ICONIC:
-
Infection response through virus genomics
- UCL:
-
University College London
- NCBI:
-
National Centre for Biotechnology Information.
References
Fischer N, Indenbirken D, Meyer T, Lutgehetmann M, Lellek H, Spohn M, et al. Evaluation of unbiased next-generation sequencing of RNA (RNA-seq) as a diagnostic method in influenza virus-positive respiratory samples. J Clin Microbiol. 2015;53(7):2238–50.
Kampmann ML, Fordyce SL, Avila-Arcos MC, Rasmussen M, Willerslev E, Nielsen LP, et al. A simple method for the parallel deep sequencing of full influenza a genomes. J Virol Methods. 2011;178(1–2):243–8.
Rutvisuttinunt W, Chinnawirotpisan P, Simasathien S, Shrestha SK, Yoon IK, Klungthong C, et al. Simultaneous and complete genome sequencing of influenza a and B with high coverage by Illumina MiSeq platform. J Virol Methods. 2013;193(2):394–404.
Zhou B, Lin X, Wang W, Halpin RA, Bera J, Stockwell TB, et al. Universal influenza B virus genomic amplification facilitates sequencing, diagnostics, and reverse genetics. J Clin Microbiol. 2014;52(5):1330–7.
Watson SJ, Welkers MR, Depledge DP, Coulter E, Breuer JM, de Jong MD, et al. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos Trans R Soc Lond Ser B Biol Sci. 2013;368(1614):20120205.
Van den Hoecke S, Verhelst J, Vuylsteke M, Saelens X. Analysis of the genetic diversity of.influenza A viruses using next-generation DNA sequencing. BMC Genomics. 2015;16:79.
Bidzhieva B, Zagorodnyaya T, Karagiannis K, Simonyan V, Laassri M, Chumakov K. Deep sequencing approach for genetic stability evaluation of influenza a viruses. J Virol Methods. 2014;199:68–75.
Kuroda M, Katano H, Nakajima N, Tobiume M, Ainai A, Sekizuka T, et al. Characterization of.quasispecies of pandemic 2009 influenza A virus (A/H1N1/2009) by de novo sequencing using a next-generation DNA sequencer. PLoS One. 2010;5(4):e10256.
Jonges M, Welkers MR, Jeeninga RE, Meijer A, Schneeberger P, Fouchier RA, et al. Emergence of the virulence-associated PB2 E627K substitution in a fatal human case of highly pathogenic avian influenza virus a(H7N7) infection as determined by Illumina ultra-deep sequencing. J Virol. 2014;88(3):1694–702.
Baum A, Sachidanandam R, Garcia-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A. 2010;107(37):16303–8.
Rudge JW, Budayanti NS, Adisasmito W, et al. Surveillance and characterisation of influenza among patients with influenza-like illness in Bali, Indonesia. Int J Infect Dis. 2012;16:e14.
WHO. Cumulative number of confirmed human cases of avian influenza A(H5N1) reported to WHO 2015 2015 [cited 2015 10 September 2015].
Simmons P. Perspectives on the 2003 and 2004 avian influenza outbreak in Bali and Lombok. Agribusiness. 2006;22(4):435–50.
Santhia K, Ramy A, Jayaningsih P, Samaan G, Putra AAG, Dibia N, et al. Avian influenza a H5N1 infections in Bali Province, Indonesia: a behavioral, virological and seroepidemiological study. Influenza Other Respir Viruses. 2009;3(3):81–9.
Harvala H, Frampton D, Grant P, Raffle J, Ferns RB, Kozlakidis Z, Kellam P, Pillay D, Hayward A, Nastouli E, For the ICONIC Consortium. Emergence of a novel subclade of influenza A(H3N2) virus in London, December 2016 to January 2017. Euro Surveill. 2017; 22(8): pii=30466.
Gallo Cassarino T., Frampton D, Sugar R, et al. High-throughput pipeline for the de novo assembly and drug resistance mutations identification from Next-Generation Sequencing viral data of residual diagnostic samples. 2016 (in preparation, bioRxiv pre-print deposition. doi: https://doi.org/10.1101/035154)
Hunt M, Gall A, Ong SH, Brener J, Ferns B, et al. IVA: accurate de novo assembly of RNA virus genomes. Bioinformatics. 2015;31(14):2374–610.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D 17 October 2007. The Influenza Virus Resource at the National Center for Biotechnology Information.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–63.
Guindon, Gascuel. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(2003):696–704.
Acknowledgements
We thank the INDOHUN university network that has consisted of 34 faculties in 20 universities in Indonesia, as an umbrella for bridging collaboration with Farr Institute University College London to help with Whole Genome Sequencing Analysis. We would like to express our appreciation to Bali Provincial Health Office for helping the data collection and 21 health facilities involved for participants recruitment including Selat Health Centre, Kintamani 1 Health centre, Klungkung 1 Health Centre, Gianyar 1 Health Centre, Denpasar Selatan Health Centre, Denpasar Barat Health Centre, Kuta 1 Health Centre, Mengwi 1 Health Centre, Penebel 1 Health Centre, Negara 2 Health Centre, Sawang 1 Health Centre, RSUD Bangli, RSUD Karangasem, RSUD Klungkung, RSUD Sanjiwani Gianyar, RSUD Tabanan, RSUD Negara, RSUD Singaraja, RSUD Kapal, RSUP Sanglah, and RSU Wangaya. We acknowledge the support from Biomolecular laboratory staff of Udayana University and Pandu Biosains for conducting the whole genome sequence of collected samples. We also thank Professor I Ngurah Mahardika for virological and laboratory advice.
Funding
The BaliMEI project is funded by Hoffman La-Roche. It is an international research collaboration between Universitas Indonesia and London School of Hygiene and Tropical Medicine (LSHTM).
Drs Gallo Cassarino, Watson and Kozlakidis were supported by the Health Innovation Challenge Fund T5–344 (ICONIC), a parallel funding partnership between the Department of Health and Wellcome Trust. The views expressed in this publication are those of the author(s) and not necessarily those of the Department of Health or Wellcome Trust. Dr. Kozlakidis was supported additionally by the UCL 2016 Knowledge Exchange Champion Award.
Availability of data and materials
The datasets generated during and/or analysed during the current study are available through the GISAID platform using the ID numbers shown in the “Additional file 2”.
Author information
Authors and Affiliations
Contributions
BSN and AND participated in the study design and coordination, collected and processed the samples and collected the associated clinical data. GCT, WSJ, RJW, SGJD, KZ and CR analyzed and interpreted the collected patient and genomic data and contributed in writing the manuscript. AW implemented the study. AW and KZ wrote the manuscript. All authors read and approved the final manuscript
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted under ethics consent, more specifically ethics approval numbers: 41/H2.F10/PPM.00/2010 (University of Indonesia) and 441/Skrt/VI/2010 (Udayana University). Written informed consent was obtained from each participating patient or their legal representatives.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Sequence IDs from the GISAID database. Sequences downloaded from the GISAID database representing the range of influenza genetic diversity worldwide during the same period that the BaliMEI study was conducted (XLS 876 kb)
Additional file 2:
Sequence IDs from the GISAID database. The IDs of the BaliMEI influenza sequences uploaded to the GISAID database (XLS 79 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Adisasmito, W., Budayanti, S.N., Aisyah, D.N. et al. Phylogenetic characterisation of circulating, clinical influenza isolates from Bali, Indonesia: preliminary report from the BaliMEI project. BMC Infect Dis 17, 583 (2017). https://doi.org/10.1186/s12879-017-2684-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-017-2684-2