
Abstract
Background
Delirium is a prevalent neuropsychiatric medical phenomenon that causes serious emergency outcomes, including mortality and morbidity. It also increases the suffering and the economic burden for families and carers. Unfortunately, the pathophysiology of delirium is still unknown, which is a major obstacle to therapeutic development. The modern network-based system biology and multi-omics analysis approach has been widely used to recover the key drug target biomolecules and signaling pathways associated with disease pathophysiology. This study aimed to identify the major drug target hub-proteins associated with delirium, their regulatory molecules with functional pathways, and repurposable drug candidates for delirium treatment.
Methods
We used a comprehensive proteomic seed dataset derived from a systematic literature review and the Comparative Toxicogenomics Database (CTD). An integrated multi-omics network-based bioinformatics approach was utilized in this study. The STRING database was used to construct the protein-protein interaction (PPI) network. The gene set enrichment and signaling pathways analysis, the regulatory transcription factors and microRNAs were conducted using delirium-associated genes. Finally, hub-proteins associated repurposable drugs were retrieved from CMap database.
Results
We have distinguished 11 drug targeted hub-proteins (MAPK1, MAPK3, TP53, JUN, STAT3, SRC, RELA, AKT1, MAPK14, HSP90AA1 and DLG4), 5 transcription factors (FOXC1, GATA2, YY1, TFAP2A and SREBF1) and 6 microRNA (miR-375, miR-17-5, miR-17-5p, miR-106a-5p, miR-125b-5p, and miR-125a-5p) associated with delirium. The functional enrichment and pathway analysis revealed the cytokines, inflammation, postoperative pain, oxidative stress-associated pathways, developmental biology, shigellosis and cellular senescence which are closely connected with delirium development and the hallmarks of aging. The hub-proteins associated computationally identified repurposable drugs were retrieved from database. The predicted drug molecules including aspirin, irbesartan, ephedrine-(racemic), nedocromil, and guanidine were characterized as anti-inflammatory, stimulating the central nervous system, neuroprotective medication based on the existing literatures. The drug molecules may play an important role for therapeutic development against delirium if they are investigated more extensively through clinical trials and various wet lab experiments.
Conclusion
This study could possibly help future research on investigating the delirium-associated therapeutic target biomarker hub-proteins and repurposed drug compounds. These results will also aid understanding of the molecular mechanisms that underlie the pathophysiology of delirium onset and molecular function.
Similar content being viewed by others


Background
Delirium is a serious neuropsychiatric medical condition triggered by multiple predisposing and precipitating factors, including critical medical situations, drug usage or withdrawal, and major surgery [1, 2]. It is a common phenomenon that increases mortality and morbidity, and prolongs hospital stays, and increases overall costs [3, 4]. Delirium causes a great deal of suffering in both carers and patients [2, 5]. More than 50% of delirious cases are undiagnosed in hospitalized patients, particularly in intensive care units (ICU) [4, 6,7,8,9]. The underlying causes for the difficulties of diagnosis reveal the deficiency of definition about delirium and ICU syndrome, other associated factors, and most importantly, the entire molecular pathophysiology of delirium [10]. The entire disease pathophysiology consists of risk markers, disease markers, end products and their combination acting in different biological processes for disease development and proliferation. The current literature suggested that various factors related to the pathophysiology of delirium have been studied, including inflammation and neuroinflammation, chronic stress, impaired blood brain barrier integrity, neuronal injury, reduced neuroprotection, cholinergic deficiency, and the effects of anticholinergic drug [11]. The pathophysiology of delirium may be understood by identifying biomarkers, molecular pathways, and predictors of delirium using multi-omics-based system biology techniques.
Delirium is prompted by distinct factors, however, the pathophysiological mechanism of delirium presentation is still unknown [2, 12, 13]. Molecular key components also play a major and significant role in delirium development, with various studies revealing the risk factors, including the genetic causes, proteins, and other biomolecules in its appearance [4, 14,15,16,17,18]. The few genetic investigations that have been conducted are powered modestly, and hence, no consistent genomic or proteomic signature candidate linked to delirium risk, diagnosis, and therapeutics have been found [1, 19, 20]. Therefore, the entire pathophysiology related to delirium has remained unknown, thus demanding an in-depth rigorous molecular investigation.
However, significant molecular biomarkers can act as risk and disease markers [21]. Modern transcriptional and proteomic data analysis provides phenomenal insight into the key molecular biomarkers and their functional pathways for specific biological traits [22,23,24,25,26]. Furthermore, integrative network-based system biology and bioinformatics approaches are widely used for molecular investigation, illuminating mysterious disease pathogenesis. In this regard, significant proteomic biomarkers effectively reveal disease severity, risk, onset, recovery, and pathway of the illness.
A recent study conducted by Takahaschi et al. 2020 [27] reported some key delirium-associated genes and their associated functional pathways using a dataset from the Comparative Toxicogenomics Database (CTD). Although the study reported some important molecular insights, it exclusively used database-derived genomic data, with the exception of one gene expression dataset. The study failed to account for the crucial pre- and post-transcriptional regulatory molecules which are very important for controlling gene expression. The study also did not provide any information about the therapeutic agents associated with delirium.
Therefore, we focused on collecting a comprehensive seed genomic dataset including available gene expression data along with the database information. This study was designed to identify the delirium-associated key proteomic biomarkers, their associated pre- and post-transcriptional regulatory molecules, and their functional pathways using an integrative bioinformatics analytical approach. Using drug repurposing techniques, the probable computational repurposable drug candidates associated with the key proteomic biomarkers were also identified. The outcome of this study will serve as a rational basis for further molecular in-depth investigation regarding the key proteomic biomarkers of delirium focusing on diagnostic and therapeutic development.
Materials and methods
Data collection
This study constructed a comprehensive proteomic dataset by combining two data sources. Firstly, a thorough review of the literature was conducted on delirium-related proteomic signatures. The electronic bibliographic databases PubMed, Scopus, and EBSCOhost (CINAHL, Medline) were utilized and searched articles between January 1, 2000, and December 31, 2022. The primary keywords were “delirium” and “biomarker” used along with a combination of other associated keywords including “marker”, ‘genetic”, “proteomic”, “genes” and “protein” to search the studies. Boolean operators “AND”, and “OR” were applied to combine the searching keywords. Fifty-four studies were included in our analysis out of a total of 2,065 that were retrieved and reviewed. The delirium-associated unique proteomic signatures from those selected studies were collected, which included 154 unique gene-encoded proteins. The detailed procedure of the literature review for this data collection has been described in Supplementary File 1.
Second, delirium-associated genes were collected from the CTD (http://ctdbase.org/) [28] which has been widely used to explore the chemical-genes/proteins interactions, gene-disease interactions as well as chemical-disease interactions. These database-retrieved interactions help researchers better understand the disease mechanism brought on by chemicals or chemical-associated genes/proteins. Delirium-related genomic data were retrieved from the CTD using the term “delirium” as the search term. The CTD inference score > 40 was considered in this study to select the top-ranking delirium-associated genes [27]. A total of 350 delirium-related genes were extracted from CTD.
The final seed dataset for this study was constructed by combining the above two datasets. If the literature-retrieved dataset is noted by A and the CTD-retrieved dataset is annotated by B, then the ultimate dataset is Z=(AUB). The following Venn diagram describes the seed dataset distribution (Fig. 1).

Venn diagram visualizing the seed dataset of this study
Methods
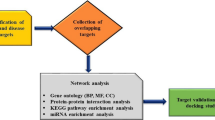
The widely used integrated bioinformatics and system biology analytic methodologies were applied in this study and are explained below. The transcriptome-guided network-based investigation employed a novel analytical technique to determine the leading biomolecules. The entire working flow diagram of this study is presented in Fig. 2.

The global working flow diagram of this study
Human protein-protein Interaction (PPI) network construction
To construct the human Protein-Protein Interaction (PPI) network, the seed dataset was utilized where the genes were mapped to the human proteins as gene-encoded proteins. To carry out a certain function, bigger protein complexes are formed with the help of the PPI network [29]. The top-ranked hub-proteins are considered the most significant proteins for biological function based on the number of connections with other nodes. PPI network analysis is one of the most effective and widely used techniques to reveal hub-proteins. In this study, the STRING database [30] has been used to construct the PPI network. PPI network topological studies and visualization were carried out using NetworkAnalyst [31] and Cytoscape 3.7.2 [31]. A topological exploration based on dual-metric measurements degree of connectivity and betweenness was utilized to identify the highly significant hub-proteins.
Physicochemical properties of hub proteins
The online project ProtParam (https://web.expasy.org/protparam/) provided the physicochemical characteristics of the hub-proteins that have been identified in this study. The server calculates numerous chemical and physical characteristics for a certain protein. For the reported hub protein, the molecular weight, theoretical pI, extinction coefficient, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) were all examined in this study.
Functional and pathway enrichment analysis
The gene set enrichment and annotation analyses [32,33,34,35] which are known as the biological processes (BP), molecular functions (MF), and cellular components (CC) and the functional and signaling pathways were carried out using the top-ranked hub-proteins. The g:GOSt software embedded in the g:Profiler web server was utilized to perform the enrichment and annotation analyses. The signaling pathways related to delirium-associated key genes were retrieved from three different databases called, KEGG, REACTOME and WIKIPATHWAYS. The statistically significant gene ontology (GO) terms and the significant pathways for hub-proteins were defined by the adjusted p-values < 0.05 and the Benjamini and Hochberg [36] procedure controlling the false discovery rate ( FDR).
Regulatory network analysis
The significant pre- and post-transcriptional regulatory molecules related to delirium-associated proteins were identified by analyzing the hub-proteins and transcription factor (TF) interaction network as well as the hub-proteins and microRNA (miRNA) interaction network, respectively. For this purpose, the JASPAR [37] TF database was utilized to construct the TF-Hub-proteins interaction network. The TarBase V8.0 and miRTarBase [38, 39] miRNAs databases were utilized to construct the interaction networks among the hub-proteins and miRNAs. The key important regulatory molecules were selected using the NetworkAnalyst online server, which applied the maximum topological matrices (degree of interconnection and betweenness) to the interaction network.
Drug-repurposing analysis for the hub-proteins
The delirium-associated hub-proteins were used to identify the repurposable drugs/drug candidates. The online drug-repositioning tool and database Connectivity Map (CMap) were used to obtain the compounds that were likely to be medications or drug candidates based on delirium-associated hub-proteins guidance [49]. This platform integrates knowledge concerning drugs or drug candidate molecules from publicly accessible sources of data in clinical experimental phases, investigative stages, and stages where they have been authorized for use in treating patients. In this study, only FDA authorized and launched drugs that are related to the delirium-associated hub-proteins were collected.
Results
Dataset description
The comprehensive literature review of delirium-associated gene-encoded proteins was collected from the finally included studies. Finally, fifty-four studies revealed a set of 154 unique proteins. In the review, the included studies were conducted in different settings and in patients with critical medical conditions. The main goal was to identify the delirium-associated genomic/proteomic signature. The well-established and widely used delirium assessment methods were used to confirm the patient’s delirious condition. To collect the gene-encoded protein dataset, we have only considered delirium-associated genes and proteins. On the other side, the CTD database was utilized to retrieve the delirium-associated genomic signatures. The database provided a search outcome of 25,036 delirium-associated genes when the lower inference score was included. After setting an inference score cutoff (> 40), the number of genes decreased to 350 genes. The ultimate seed dataset was constructed by combining the two datasets from different sources, consisting of 477 unique gene-encoded proteins (Supplementary file 2). Between the two datasets, 323 unique proteins come from set A and 127 come from set B and 27 genes were found to be in common. Therefore, for the downstream analysis, 477 genes/proteins were utilized in this study.
PPI network analysis
The PPI network of the 477 collected gene encoded proteins, was constructed using the STRING database (Fig. 3). Among the 477 proteins, 4 proteins were not detected by STRING database, hence they were excluded from the network. A dual-metric topological measurement of the degree of connectivity and the betweenness were considered to identify the central highly connected representative hub-proteins. The key 11 hub-proteins were identified based on their degree and betweenness using topological analysis measured by the CytoHubba. The hub-proteins are MAPK1, MAPK3, TP53, JUN, STAT3, SRC, RELA, AKT1, MAPK14, HSP90AA1 and DLG4. The hub-proteins are highlighted in Fig. 3. These hub-proteins were different compared to those in the previously conducted study by Takahaschi et al. 2020 [27].

PPI network of delirium-associated proteins. The smaller dark blue circular nodes represent the network proteins. The bigger circular colorful nodes represent the hub-proteins where a redder color node means a higher degree of connectivity and a more yellow color node means a lower degree of connectivity measured by CytoHubba
This study reports the physicochemical characteristics of the detected delirium-associated hub-proteins. The physicochemical properties are necessary for a more thorough analysis of the reported key biomolecules. The number of amino acids varied from 331 (JUN) to 770 (STAT3) among the hub-proteins when they also have the lowest (35675.57 kda) and highest (88067.80 kda) molecular weight, respectively. The theoretical isoelectric point (pI) revealed that the hub-protein JUN consisted of the highest pI value of 8.90 and the lowest pI value of 4.94 was contained in the HSP90AA1 hub-protein. Detailed properties information of the hub-proteins is provided in Table 1.
Functional annotation and enrichment analysis
The gene ontology (GO) functional enrichment and annotation analysis of the hub-proteins are represented in three categories, namely the BP, MF, and CC. In Fig. 4A, the top ten significant and enriched GO terms from each of the three categories (BP, MF, and CC) have been summarized. The top ten functional signaling pathways of the hub-proteins from three different databases are reported in Fig. 4B. The GO functional enrichment and pathway analysis of the delirium-associated hub-proteins revealed a wide range of biological and functional pathways. Among the significant BPs, the response to organonitrogen compound, response to growth factor, cellular response to biotic stimulus, response to oxidative stress, cellular response to reactive oxygen species, cellular response to chemical stress, and regulation of signal transduction were highly considerable. The highly enriched and significant MFs were enzyme binding, protein kinase binding, phosphatase binding, kinase binding, and protein serine/threonine/tyrosine kinase activity. Moreover, glutamatergic synapse, nucleoplasm, cytosol, synapse, and nuclear lumen were the hub-proteins’ highly enriched and significant cellular locations (Fig. 4A).

(A) The top ten significant GO terms associated with the biological process (BP), molecular function (MF) and cellular component (CC) has been presented. (B)The three-database retrieved top significant signaling pathways shared by delirium-associated key genes have been presented. In both figures, the size of the bubbles indicates the number of key genes enriched in each term
On the other hand, the noticeable significant signaling pathways of the hub-proteins mainly consisted of receptor activities, stress and immune responses, cytokines and inflammation, and signaling and developmental pathways. The signaling pathways were those that Takahaschi et al. 2020 [27] identified in relation to delirium. The significant functional pathways shared by the hub-proteins were lipid and atherosclerosis, cell signal transduction, RAC1/PAK1/p38/MMP2 pathway, AGE/RAGE pathway, cytokine signaling pathway, developmental biology, shigellosis, cellular senescence, signaling by interleukins and other receptor associated pathways (Fig. 4B). The significantly enriched pathways are associated with the components of hallmarks of aging [40]. The details of functional enrichment and pathway analysis results are provided in Supplementary File 3.
Identification of hub-proteins-related regulatory factors
The pre- and post-transcriptional regulatory molecules of the reported therapeutic target, hub-proteins, have been identified throughout the interaction network analysis. The interaction network among the drug-targeted proteins revealed the key TFs were, namely, FOXC1, GATA2, YY1, TFAP2A, and SREBF1 based on the topological analysis by CytoHubba (Fig. 5A). Similar topological measurements of degree and betweenness centrality were applied to the interaction network of miRNAs and hub proteins. The network analysis from the two databases showed similar results for the post-transcriptional regulatory miRNAs. The substantial top regulatory miRNAs were recorded as miR-375, miR-17-5, miR-17-5p, miR-106a-5p, miR-125b-5p, and miR-125a-5p (Fig. 5B C). Consistency among regulatory elements from different databases revealed the significance of these molecules for hub proteins in delirium.

(A) TFs and the hub-proteins interaction network were constructed from the JASPAR database The pink-colored octagonal nodes represent the key TFs while the blue color triangle nodes are for hub-proteins and the green color nodes stand for the associated TFs. The miRNAs versus hub-proteins interaction network (B) were constructed from the TarBase, whereas the second network (C) was built using the miTarBase database. In both networks, the dark, blue-colored hexagonal nodes represent the key miRNAs. The yellow-colored circular nodes stand for hub-proteins and the light, blue-colored circular nodes represent the other associated miRNAs
Drug-repurposing
The integrative comprehensive drug database CMap revealed a total of 17 FDA approved drug molecules targeting our proposed delirium-associated hub-proteins. Among the retrieved drugs, 11 of them are inhibitors, four were receptor drugs, one is a stimulant, and one is a mucolytic agent (Table 2). The only hub-protein, MAPK14 did not retrieve any associated repurposable drugs from the CMap database. Although all the hub-proteins also found many other repurposable drugs that are still in different clinical trial stages. However, these were ignored and not reported.
The repurposable drugs have shown effectiveness across different disease areas, including neurology/psychiatry, nephrology, cardiology, gastroenterology, various oncology, and others (Table 2). These computationally predicted drugs need deeper clinical investigation before serving as a potential treatment source for delirium.
Discussion
The literature review showed that very few proteomic-level studies have been conducted to identify the key proteins associated with delirium [1, 19, 20]. Nevertheless, to the best of our knowledge, we were unable to locate any gene expression data regarding delirium. Due to the paucity of gene expression transcriptomics data, this study planned to collect a distinct molecular dataset focusing on the genes and proteins associated with delirium. Accordingly, a comprehensive systematic review of 2,065 articles revealed 54 studies that reported 154 unique delirium-associated gene-encoded proteins, although none of them were consistent across studies. On the other side, the CTD database provided a list of 350 gene-encoded proteins which assisted to make a comprehensive proteomic seed dataset of 477 unique gene-encoded proteins that could be analyzed. The current seed dataset contains more delirium-associated genomic indicators which were reported by high throughput gene expression data analysis under few targeted genes/proteins in different studies. This dataset diversity and its settings difference can explain the delirium-associated genes from different biological aspects which is highly significant and thus distinguishes this study from others. Based on this dataset, the current study aimed to identify the key proteomic biomarkers, their regulatory molecules, the functional pathways, and the repurposable drug components associated with delirium-associated hub-proteins. Using the network-based multi-omics data integration framework analysis approach, the study was able to identify the delirium-associated key pre-clinical substantial drug target biomolecules. This study outcome focused on highlighting the pathophysiology of delirium focused on the molecular functionality aspects.
The integrated bioinformatics analysis approach revealed 11 hub-proteins (MAPK1, MAPK3, TP53, JUN, STAT3, SRC, RELA, AKT1, MAPK14, HSP90AA1 and DLG4), 5 TFs (FOXC1, GATA2, YY1, TFAP2A and SREBF1) and 6 miRNAs (miR-375, miR-17-5, miR-17-5p, miR-106a-5p, miR-125b-5p, and miR-125a-5p) associated with delirium. The PPI network revealed that the hub-proteins differed from those of the other study [27] because of data diversity and study settings. This analytical dataset contained genes collected from different studies which were in distinct settings using biological samples of the human body. The system biology approach has the privilege of accumulating genomic information through the network-based analysis which reflects the global key hub-proteins for a specific disease. Among the hub-proteins, the mitogen-activated protein kinase 1 (MAPK1) proteins and its mutants are associated with neurodevelopmental disorders, which eventually phenotype with several neurological challenges including intellectual disability, growing delay, and interactive difficulties such as anxiety, reduced stress tolerance and aggressive behaviour [41]. In addition to pro-inflammatory stimuli, cellular and environmental stressors also activate the MAPK signaling pathways [42, 43], whereas delirious conditions are also triggered by the stressed medical condition and inflammatory cytokines activities [4, 44, 45]. The reported MAPK signaling pathway-related hub proteins, namely MAPK1, MAPK3 and MAPK14, might be involved in delirium-associated neurological disorders/activities which demand deeper investigation. The tumor suppressor gene, TP53 encoded proteins, is also known as TP53/p53, is a crucial protein in various cancer and tumor development [46,47,48]. A wide interaction with cytokine and chemokines inflammatory components is revealed by the p53 pathway and other family members [49], indicating a positive association with delirium development under critical and stressful medical conditions. The c-Jun type gene encoded proteins JUN has a greater involvement in different cell activities, disease proliferation, and stress-response signaling pathways [50,51,52]. The ischemic/reperfusion consequences in the perioperative period and the postoperative pain sedation are activated by the JAK/STAT3 signaling pathway with the assistance of other inflammatory cytokines (like, IL6). The research revealed that postoperative pain and inflammation are major causes of delirium [49,50,51], indicating a strong association of the above proteins with delirium phases. The SRC protein is crucial in chronic inflammation and cancer development [53]. The RELA protein is treated as a potential cancer biomarker, responsible for cytokine production and inflammatory bowel disease [54, 55], when AKT1 is a protein that aids in the correct growth and operation of the nervous system [56,57,58]. The HSP90AA1 gene-encoded protein’s expression is associated with the inflammatory protein interleukins (IL) and the regulatory role in neurodevelopment, indicating a core connection with cognitive dysfunction and delirium [59, 60]. The hub protein DLG4 is involved in the microglial inflammatory process, and genetic differences in DLG4 are linked to anatomical variations in the preterm newborn brain [61]. The above hub-proteins showed a diverse functionality related to delirium, other neural dysfunction and cognitive impairment. The linkage clearly indicates the significance of the hub-proteins in delirium-associated functional pathways and pathophysiology. Most of them revealed cytokines inflammation, neurological disorders/activities, stress responsiveness, and pain management, which are parallel causes of delirium in acute medical conditions.
Functional enrichment and signaling pathways analysis is one of the most effective ways to decipher a group of genes’ molecular activities and functional pathways. Among the GO terms, the cellular response to oxidative stress is an important pathway associated with chronic inflammation and aged diseases [62] which is enriched by seven hub-proteins. Oxidative stress might be associated with delirium pathophysiology as it is predominantly observed among older patients. One of the important MAPK signaling pathways was regulated by the reactive oxygen species (ROS) biological function induced by oxidative stressors [63]. The hub proteins response to chemical stress is an important part of many pathogenesis and neurodegenerative diseases and inflammation [64]. The drugs and medications used for anesthesia and surgery are involved in delirium under the chemical stress response. Among the MFs, most of the hub proteins are involved in kinase activities. In response to a wide range of stimuli, including mitogens, stress, heat shock, and proinflammatory cytokines, MAPKs activities control cellular responses [65, 66]. The MAPK signaling pathways might be crucial for delirium pathophysiology since the key delirium-associated hub proteins MAPK1, MAPK3, and MAPK14 were found to be associated with it. Besides that, the cellular senescence, lipid and atherosclerosis, shigellosis, developmental biology, various receptor associated pathways, cytokines and interleukins associated pathways and stress and inflammation related pathways were significantly shared by the delirium-associated hub-proteins. The shared signaling pathways consisted of the receptor-associated pathways reported by Takahaschi et al. 2020 [27]. ]. Furthermore, the current study found the stress and inflammation associated pathways shared by the hub-proteins which are also supported by the existing literature [4, 27, 67]. The GO analysis and the signaling pathway analysis revealed significant information about the molecular mechanism of delirium-associated hub-proteins that are essentially important for delirium-related molecular and pathophysiological knowledge.
The TFs and the miRNAs play significant roles in the protein translation from a specific gene. The hub-proteins are also regulated by the key transcriptional and post-transcriptional regulatory molecules that have been detected in this study. The TF FOXC1 is associated with neuroinflammation and neuronal apoptosis, whereas neuroinflammation is highly related to neurodegenerative complications such as Alzheimer’s disease, Dementia, and Parkinson’s disease [68,69,70]. Neuroglobin (NGB) gene expression is associated with neural disease (Alzheimer’s Disease) when the GATA2 TF works to regulate the NGB gene expression [71]. Studies suggest that the Yin Yang 1 (YY1) TF correlates with the central nervous system. The YY1 regulates a significant number of genes associated with the nervous system [72], hence it might have greater involvement with delirium pathophysiology. The Activator Protein 2 (AP-2) transcription factor (TF) family has a vital involvement in gene expression regulation and various cancers development with the other members in this family [73]. The SREBF1 TF is significantly associated with neuropsychiatric disorder schizophrenia [74]. On the other hand, among the post-transcriptional regulatory element miRNAs, the overexpression of miR-375 is associated with Alzheimer’s and Parkinson’s disease [75, 76] with the miR-17-5 and miR-17-5p being related to various cancer developments [3, 77, 78]. The miR-106a-5p miRNA, has a strong connection to the regulation of CD4 + T-cells and functions as a tumor suppressor, and miR-125b-5p is interrelated with suppressing PI3K/AKT pathway in bladder cancer [79] and the miR-125a-5p is connected with the macrophages inflammatory response [80].
The reported hub-proteins and their signaling pathways, TFs were also associated with the hallmarks of aging components since age is considered one of the most important and significant factors for delirium. Among the nine hallmarks of aging [40] the cellular senescence, loss of proteostasis, alteration in intercellular communication, genomic and epigenomic alteration are closely connected with the reported hub-protein’s functions and enriched signaling pathways. For example, the cellular senescence signaling pathway was significantly enriched by the eight hub-proteins. The cytokines and inflammatory related pathways, signal transduction, oxidative stress and telomerase activity are significant signaling pathways closely connected with the loss of proteostasis, alteration in intercellular communication hallmarks of aging.
The hub-proteins related computationally identified repurposable drugs were retrieved from the database. The predicted drug molecules including aspirin, irbesartan, ephedrine-(racemic), nedocromil, and guanidine were characterized as anti-inflammatory, stimulating the central nervous system, and neuroprotective medication as confirmed by the existing literature [81,82,83,84,85,86]. The drug molecules may play an important role for therapeutic development against delirium if they are investigated more extensively though clinical trials and various wet lab experiments. Our results therefore provide a platform for future investigations into the underlying mechanisms of delirium and therapeutic treatment development.
The overall discussion about the key hub-proteins and their regulatory molecules revealed that the reported biomarkers have an agglomerative and strong interconnection with the delirium-associated genes. The signaling pathway analysis highlighted the cytokines and inflammatory pathways, cellular senescence pathway, stress and telomerase activity pathways were significantly associated with the molecular pathophysiological mechanism of delirium. The reported drug target hub-proteins and their associated computationally identified repurposable drug molecules may open a new and extensive research dimension in the field of delirium drug development.
Implications of this study
The implications of this study are far-reaching. Firstly, the results provide a comprehensive overview of the key drug target proteins associated with delirium, their associated regulatory molecules, and functional pathways. This allows for better understanding of the molecular mechanisms underlying delirium and for a better evaluation of drug candidates for the treatment of this disorder. Secondly, the meta-data analyses of existing evidence indicate the potential of new drug targets, which could be explored further for the development of novel therapeutics. Finally, the study provides a platform for future research, allowing for further investigations into the underlying mechanisms of delirium and the development of more effective treatments. If proven, the findings from this study could be used to inform clinical practice and public health policy concerning the prevention and management of delirium.
Limitations of this study
Firstly, the study used a comprehensive seed proteomic dataset from a systematic literature review and CTD database. In both cases, the research team and the database have ensured the integrity of the genomic/proteomic information. Secondly, during the proteomic data collection, only association with delirium was considered and the magnitude of association (either positive or negative) was ignored in both sources. In this aspect, it is unknown whether the reported hub proteins are associated with delirium in their upregulated or downregulated condition. On the other hand, no rigorous study has been found that reported the comprehensive gene expression data or any transcriptomics dataset which could reveal up and downregulated genes/proteins associated with delirium. Moreover, the meta-data used in the study may not be comprehensive or up to date. The study may be limited by potential confounding factors that may influence the association between drug target proteins, regulatory molecules and functional pathways and the development of delirium.
Conclusion
Since delirium is considered a multifactorial critical medical condition, its anatomic molecular functions and pathophysiology might be more diverse and complicated. The ongoing genomic research and studies revealed some snapshots of the disease’s molecular diversity. A wide range of transcriptomics data can be generated and analyzed to identify the important genes at the critical time point of delirious patients, revealing the differentially expressed genes and proteins. More studies should be undertaken to collect gene expression data and hence map out their functional pathways and molecular mechanisms rigorously. The current study carried out a multi-omics network-based system biology analysis using the comprehensive delirium associated genomic dataset. The study revealed the key hub-proteins on a highly interconnected PPI network. The signaling pathways analysis of the hub-proteins showed different significant pathways associated with delirium and the hallmarks of aging. The study also identified some important regulatory molecules linked to delirium associated hub-proteins. The biomolecules identified in this study may have potential association with delirium which will contribute to future research by enhancing our understanding of the delirium mechanism. Moreover, the computationally predicted repurposable drug molecules associated with the hub-proteins as reported in this study provide a wide range of research opportunities in therapeutic developments against delirium. The study will also provide a platform for future investigations into the underlying mechanisms of delirium.
Data Availability
All data generated or analyzed during this study are either publicly available or included in this article.
References
Wilson JE, Mart MF, Cunningham C, Shehabi Y, Girard TD, MacLullich AMJ, et al. Delirium Nat Rev Dis Prim. 2020;6:90. https://doi.org/10.1038/s41572-020-00223-4.
Toft K, Tontsch J, Abdelhamid S, Steiner L, Siegemund M, Hollinger A. Serum biomarkers of delirium in the elderly: a narrative review. Ann Intensive Care. 2019;9:76. https://doi.org/10.1186/s13613-019-0548-1.
Mosharaf MP, Kibria MK, Hossen MB, Islam MA, Reza MS, Mahumud RA, et al. Meta-Data Analysis to explore the hub of the hub-genes that influence SARS-CoV-2 Infections highlighting their pathogenetic processes and Drugs repurposing. Vaccines. 2022;10:1248. https://doi.org/10.3390/vaccines10081248.
Dunne SS, Coffey JC, Konje S, Gasior S, Clancy CC, Gulati G, et al. Biomarkers in delirium: a systematic review. J Psychosom Res. 2021;147:110530. https://doi.org/10.1016/j.jpsychores.2021.110530.
Williams ST, Dhesi JK, Partridge JSL. Distress in delirium: causes, assessment and management. Eur Geriatr Med. 2020;11:63–70. https://doi.org/10.1007/s41999-019-00276-z.
Mikhailovich A. The American geriatrics society/national institute on aging bedside-to-bench conference: Research agenda on delirium in older adults. J Am Geriatr Soc. 2015;63:843–52. https://doi.org/10.1111/jgs.13406.
Marcantonio ER. Delirium in hospitalized older adults. N Engl J Med. 2017;377:1456–66. https://doi.org/10.1056/NEJMcp1605501.
Poulsen LM, Estrup S, Mortensen CB, Andersen-Ranberg NC. Delirium in Intensive Care. Curr Anesthesiol Rep. 2021;11:516–23. https://doi.org/10.1007/s40140-021-00476-z.
Han JH, Zimmerman EE, Cutler N, Schnelle J, Morandi A, Dittus RS, et al. Delirium in older emergency department patients: Recognition, risk factors, and psychomotor subtypes. Acad Emerg Med. 2009;16:193–200. https://doi.org/10.1111/j.1553-2712.2008.00339.x.
Sepulveda E, Franco JG, Trzepacz PT, Gaviria AM, Meagher DJ, Palma J, et al. Delirium diagnosis defined by cluster analysis of symptoms versus diagnosis by DSM and ICD criteria: diagnostic accuracy study. BMC Psychiatry. 2016;16:167. https://doi.org/10.1186/s12888-016-0878-6.
Vasunilashorn SM, Dillon ST, Marcantonio ER, Libermann TA. Application of multiple omics to Understand Postoperative Delirium Pathophysiology in humans. Gerontology. 2023. https://doi.org/10.1159/000533789.
Hshieh TT, Fong TG, Marcantonio ER, Inouye SK. Cholinergic deficiency hypothesis in delirium: a synthesis of current evidence. J Gerontol A Biol Sci Med Sci. 2008;63:764–72. https://doi.org/10.1093/gerona/63.7.764.
Plaschke K, Fichtenkamm P, Schramm C, Hauth S, Martin E, Verch M, et al. Early postoperative delirium after open-heart cardiac Surgery is associated with decreased bispectral EEG and increased cortisol and interleukin-6. Intensive Care Med. 2010;36:2081–9. https://doi.org/10.1007/s00134-010-2004-4.
Heinrich M, Sieg M, Kruppa J, Nürnberg P, Schreier PH, Heilmann-Heimbach S, et al. Association between genetic variants of the cholinergic system and postoperative delirium and cognitive dysfunction in elderly patients. BMC Med Genomics. 2021;14:248. https://doi.org/10.1186/s12920-021-01071-1.
Ayob F, Lam E, Ho G, Chung F, El-Beheiry H, Wong J. Pre-operative biomarkers and imaging tests as predictors of post-operative delirium in non-cardiac surgical patients: a systematic review. BMC Anesthesiol. 2019;19:25. https://doi.org/10.1186/s12871-019-0693-y.
Lindblom RPF, Shen Q, Axén S, Landegren U, Kamali-Moghaddam M, Thelin S. Protein profiling in serum and cerebrospinal fluid following complex Surgery on the thoracic aorta identifies biological markers of neurologic Injury. J Cardiovasc Transl Res. 2018;11:503–16. https://doi.org/10.1007/s12265-018-9835-8.
Hall RJ, Watne LO, Cunningham E, Zetterberg H, Shenkin SD, Wyller TB, et al. CSF biomarkers in delirium: a systematic review. Int J Geriatr Psychiatry. 2018;33:1479–500. https://doi.org/10.1002/gps.4720.
Hansen N, Krasiuk I, Titsch T. Neural autoantibodies in delirium. J Autoimmun. 2021;125:102740. https://doi.org/10.1016/j.jaut.2021.102740.
McCoy TH, Hart K, Pellegrini A, Perlis RH. Genome-wide association identifies a novel locus for delirium risk. Neurobiol Aging. 2018;68. :160.e9-160.e14.
Adamis D, Meagher D, Williams J, Mulligan O, McCarthy G. A systematic review and meta-analysis of the association between the apolipoprotein E genotype and delirium. Psychiatr Genet. 2016;26:53–9. https://doi.org/10.1097/YPG.0000000000000122.
Marcantonio ER, Rudolph JL, Culley D, Crosby G, Alsop D, Inouye SK. Serum biomarkers for delirium. J Gerontol A Biol Sci Med Sci. 2006;61:1281–6. https://doi.org/10.1093/gerona/61.12.1281.
Mosharaf MP, Reza MS, Gov E, Mahumud RA, Mollah MNH. Disclosing potential key genes, therapeutic targets and agents for Non-small Cell Lung Cancer: evidence from Integrative Bioinformatics Analysis. Vaccines. 2022;10:771. https://doi.org/10.3390/vaccines10050771.
Mosharaf MP, Reza MS, Kibria MK, Ahmed FF, Kabir MH, Hasan S, et al. Computational identification of host genomic biomarkers highlighting their functions, pathways and regulators that influence SARS-CoV-2 Infections and drug repurposing. Sci Rep. 2022;12:4279. https://doi.org/10.1038/s41598-022-08073-8.
Reza MS, Harun-Or-Roshid M, Islam MA, Hossen MA, Hossain MT, Feng S et al. Bioinformatics Screening of Potential Biomarkers from mRNA Expression Profiles to Discover Drug Targets and Agents for Cervical Cancer. Int J Mol Sci 2022, Vol 23, Page 3968. 2022;23:3968. https://doi.org/10.3390/IJMS23073968.
Moni MA, Islam MB, Rahman MR, Rashed-Al-Mahfuz M, Awal MA, Islam SMS et al. Network-Based Computational Approach to identify delineating common cell pathways influencing type 2 Diabetes and Diseases of bone and joints. IEEE Access. 2020;8.
Satu S, Khan I, Rahman R, Howlader KC, Roy S, Roy SS et al. Diseasome and comorbidities complexities of SARS-CoV-2 Infection with common malignant Diseases. Brief Bioinform. 2021;22.
Takahashi Y, Terada T, Muto Y. Systems Level Analysis and Identification of Pathways and Key genes Associated with Delirium. Genes (Basel). 2020;11:1225. https://doi.org/10.3390/genes11101225.
Davis AP, Wiegers TC, Johnson RJ, Sciaky D, Wiegers J, Mattingly CJ. Comparative toxicogenomics database (CTD): update 2023. Nucleic Acids Res. 2023;51:D1257–62. https://doi.org/10.1093/nar/gkac833.
Braun P, Gingras A-C. History of protein-protein interactions: from egg-white to complex networks. Proteomics. 2012;12:1478–98. https://doi.org/10.1002/pmic.201100563.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–8. https://doi.org/10.1093/nar/gkw937.
Xia J, Gill EE, Hancock REW. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat Protoc. 2015;10:823–44. https://doi.org/10.1038/nprot.2015.052.
Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, et al. GO::TermFinder-open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes INTRODUCTION: MOTIVATION AND DESIGN. Bioinforma Appl NOTE. 2004;20:3710–55. https://doi.org/10.1093/bioinformatics/bth456.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, Diseases and Drugs. Nucleic Acids Res. 2017;45:D353–61. https://doi.org/10.1093/nar/gkw1092.
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–62.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. https://doi.org/10.1093/nar/28.1.27.
Benjamini Y, Hochberg Y. Controlling the false Discovery rate: a practical and powerful Approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. https://doi.org/10.1111/j.2517-6161.1995.tb02031.x.
Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018;46:D260–6. https://doi.org/10.1093/nar/gkx1126.
Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, Vlachos IS, Tastsoglou S, Kanellos I, et al. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018;46:D239–45. https://doi.org/10.1093/nar/gkx1141.
Chou CH, Chang NW, Shrestha S, Hsu S, Da, Lin YL, Lee WH, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;44:D239–47. https://doi.org/10.1093/nar/gkv1258.
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039.
Motta M, Pannone L, Pantaleoni F, Bocchinfuso G, Radio FC, Cecchetti S, et al. Enhanced MAPK1 function causes a neurodevelopmental disorder within the RASopathy Clinical Spectrum. Am J Hum Genet. 2020;107:499. https://doi.org/10.1016/J.AJHG.2020.06.018.
Kyriakis JM, Avruch J. Mammalian MAPK Signal Transduction Pathways activated by stress and inflammation: a 10-Year update. Physiol Rev. 2012;92:689–737. https://doi.org/10.1152/physrev.00028.2011.
Soares-Silva M, Diniz FF, Gomes GN, Bahia D. The Mitogen-Activated Protein Kinase (MAPK) Pathway: Role in Immune Evasion by Trypanosomatids. Front Microbiol. 2016;7 FEB:183. https://doi.org/10.3389/fmicb.2016.00183.
Cerejeira JMS, Nogueira V, Luís P, Vaz-Serra A, Mukaetova-Ladinska EB. The cholinergic system and inflammation: common pathways in delirium pathophysiology. J Am Geriatr Soc. 2012;60:669–75. https://doi.org/10.1111/j.1532-5415.2011.03883.x.
Cerejeira J, Batista P, Nogueira V, Vaz-Serra A, Mukaetova-Ladinska EB. The stress response to Surgery and postoperative delirium: evidence of hypothalamic-pituitary-adrenal axis hyperresponsiveness and decreased suppression of the GH/IGF-1 Axis. J Geriatr Psychiatry Neurol. 2013;26:185–94. https://doi.org/10.1177/0891988713495449.
Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of Liver cancer. Oncogene. 2007;26:2166–76. https://doi.org/10.1038/sj.onc.1210279.
Mahumud RA, Shahjalal M. The emerging burden of genetic instability and mutation in Melanoma: role of Molecular mechanisms. Cancers (Basel). 2022;14:6202. https://doi.org/10.3390/cancers14246202.
Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hosteller R, Cleary K, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342:705–8.
Cooks T, Harris CC, Oren M. Caught in the cross Fire: p53 in inflammation. Carcinogenesis. 2014;35:1680–90. https://doi.org/10.1093/CARCIN/BGU134.
Meng Q, Xia Y. c-Jun, at the crossroad of the signaling network. Protein Cell. 2011;2:889–98. https://doi.org/10.1007/s13238-011-1113-3.
Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63:218–42. https://doi.org/10.1124/pr.110.002980.
Si Y, Zhang Y, Han L, Chen L, Xu Y, Sun F, et al. Dexmedetomidine acts via the JAK2/STAT3 pathway to attenuate isoflurane-induced neurocognitive deficits in senile mice. PLoS ONE. 2016;11:e0164763. https://doi.org/10.1371/journal.pone.0164763.
Liu ST, Pham H, Pandol SJ, Ptasznik A. Src as the link between inflammation and cancer. Front Physiol. 2014;4. https://doi.org/10.3389/fphys.2013.00416.
Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–34. https://doi.org/10.1038/NRI910.
Onishi S, Yamasaki F, Nakano Y, Takayasu T, Amatya VJ, Kolakshyapati M, et al. RELA fusion-positive anaplastic ependymoma: molecular characterization and advanced MR imaging. Brain Tumor Pathol. 2018;35:41–5. https://doi.org/10.1007/S10014-017-0301-0.
Emamian ES. AKT/GSK3 signaling pathway and schizophrenia. Front Mol Neurosci. 2012;5 MARCH:33. https://doi.org/10.3389/fnmol.2012.00033.
Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, et al. A Mosaic Activating Mutation in AKT1 Associated with the Proteus Syndrome. N Engl J Med. 2011;365:611–9. https://doi.org/10.1056/nejmoa1104017.
Schwab SG, Hoefgen B, Hanses C, Hassenbach MB, Albus M, Lerer B, et al. Further evidence for association of variants in the AKT1 gene with schizophrenia in a sample of European sib-pair families. Biol Psychiatry. 2005;58:446–50. https://doi.org/10.1016/j.biopsych.2005.05.005.
Zuehlke AD, Beebe K, Neckers L, Prince T. Regulation and function of the human HSP90AA1 gene. Gene. 2015;570:8–16. https://doi.org/10.1016/j.gene.2015.06.018.
Miller DJ, Fort PE. Heat Shock Proteins Regulatory Role in Neurodevelopment. Front Neurosci. 2018;12 NOV. https://doi.org/10.3389/fnins.2018.00821.
Krishnan ML, Van Steenwinckel J, Schang A-L, Yan J, Arnadottir J, Le Charpentier T, et al. Integrative genomics of microglia implicates DLG4 (PSD95) in the white matter development of preterm infants. Nat Commun. 2017;8:428. https://doi.org/10.1038/s41467-017-00422-w.
Khansari N, Shakiba Y, Mahmoudi M. Chronic inflammation and oxidative stress as a major cause of age-related Diseases and cancer. Recent Pat Inflamm Allergy Drug Discov. 2009;3:73–80. https://doi.org/10.2174/187221309787158371.
KrishnaMurthy A, Rathinasabapathi B. Oxidative stress tolerance in plants: novel interplay between auxin and reactive oxygen species signaling. Plant Signal Behav. 2013;8. https://doi.org/10.4161/psb.25761.
Nguyen T, Yang CS, Pickett CB. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic Biol Med. 2004;37:433–41. https://doi.org/10.1016/j.freeradbiomed.2004.04.033.
Pearson G, Robinson F, Beers Gibson T, Xu B, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–83. https://doi.org/10.1210/EDRV.22.2.0428.
Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-Activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. https://doi.org/10.1128/mmbr.00031-10.
Khan BA, Perkins AJ, Prasad NK, Shekhar A, Campbell NL, Gao S, et al. Biomarkers of Delirium Duration and Delirium Severity in the ICU. Crit Care Med. 2020;48:353–61. https://doi.org/10.1097/CCM.0000000000004139.
Wang H, Wang H, Song Y, Liu C, Qian X, Zhang D, et al. Overexpression of Foxc1 ameliorates sepsis–associated encephalopathy by inhibiting microglial migration and neuroinflammation through the IκBα/NF–κB pathway. Mol Med Rep. 2022;25:107. https://doi.org/10.3892/mmr.2022.12623.
Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s Disease: current evidence and future directions. Alzheimer’s and Dementia. 2016;12:719–32. https://doi.org/10.1016/j.jalz.2016.02.010.
Maiese K. Forkhead transcription factors: new considerations for alzheimer’s Disease and Dementia. J Transl Sci. 2016;2:241–7. https://doi.org/10.15761/jts.1000146.
Tam KT, Chan PK, Zhang W, Law PP, Tian Z, Chan GCF, et al. Identification of a novel distal regulatory element of the human neuroglobin gene by the chromosome conformation capture approach. Nucleic Acids Res. 2017;45:115–26. https://doi.org/10.1093/nar/gkw820.
He Y, Casaccia-Bonnefil P. The Yin and Yang of YY1 in the nervous system. J Neurochem. 2008;106:1493–502. https://doi.org/10.1111/j.1471-4159.2008.05486.x.
Kołat D, Kałuzińska Ż, Bednarek AK, Płuciennik E. The biological characteristics of transcription factors AP-2α and AP-2γ and their importance in various types of cancers. Biosci Rep. 2019;39. https://doi.org/10.1042/BSR20181928.
Chen Y, Bang S, McMullen MF, Kazi H, Talbot K, Ho MX, et al. Neuronal activity Induced sterol Regulatory element binding Protein-1 (SREBP1) is disrupted in dysbindin null mice – potential link to cognitive impairment in Schizophrenia. Mol Neurobiol. 2017;54:1699. https://doi.org/10.1007/S12035-016-9773-X.
Wang Q, Ge X, Zhang J, Chen L. Effect of lncRNA WT1-AS regulating WT1 on oxidative stress injury and apoptosis of neurons in Alzheimer’s Disease via inhibition of the miR-375/SIX4 axis. Aging. 2020;12:23974–95. https://doi.org/10.18632/aging.104079.
Cai L-J, Tu L, Li T, Yang X-L, Ren Y-P, Gu R et al. Up-regulation of microRNA-375 ameliorates the damage of dopaminergic neurons, reduces oxidative stress and inflammation in Parkinson’s Disease by inhibiting SP1. Aging (Albany NY). 2020;12:672–89. https://doi.org/10.18632/aging.102649.
Saral MA, Tuncer SB, Odemis DA, Erdogan OS, Erciyas SK, Saip P, et al. New biomarkers in peripheral blood of patients with Ovarian cancer: high expression levels of miR-16-5p, miR-17-5p, and miR-638. Arch Gynecol Obstet. 2022;305:193–201. https://doi.org/10.1007/S00404-021-06138-Z/TABLES/2.
Kong W, Cheng Y, Liang H, Chen Q, Xiao C, Li K, et al. Prognostic value of mir-17-5p in cancers: a meta-analysis. Onco Targets Ther. 2018;11:3541–9. https://doi.org/10.2147/OTT.S150340.
Liu S, Chen Q, Wang Y. MiR-125b-5p suppresses the Bladder cancer progression via targeting HK2 and suppressing PI3K/AKT pathway. Hum Cell. 2020;33:185–94. https://doi.org/10.1007/S13577-019-00285-X/FIGURES/4.
Banerjee S, Cui H, Xie N, Tan Z, Yang S, Icyuz M, et al. MiR-125a-5p regulates differential activation of macrophages and inflammation. J Biol Chem. 2013;288:35428–36. https://doi.org/10.1074/jbc.M112.426866.
Berk M, Dean O, Drexhage H, McNeil JJ, Moylan S, O’Neil A, et al. Aspirin: a review of its neurobiological properties and therapeutic potential for mental Illness. BMC Med. 2013;11:1–17. https://doi.org/10.1186/1741-7015-11-74/PEER-REVIEW.
Tsukuda K, Mogi M, Iwanami J, Min LJ, Jing F, Oshima K, et al. Irbesartan attenuates ischemic brain damage by inhibition of MCP-1/CCR2 signaling pathway beyond AT1 receptor blockade. Biochem Biophys Res Commun. 2011;409:275–9. https://doi.org/10.1016/J.BBRC.2011.04.142.
Indra I, Adhiany E. Medicines used in emergency. Br Int Exact Sci J. 2020;2:510–21. https://doi.org/10.33258/bioex.v2i2.227.
Alotaibi MR, Monier M, Elsayed NH. Enantiomeric resolution of ephedrine racemic mixture using molecularly imprinted carboxylic acid functionalized resin. Eur Polym J. 2019;121:109309. https://doi.org/10.1016/j.eurpolymj.2019.109309.
Gehlot P, Kumar S, Kumar Vyas V, Singh Choudhary B, Sharma M, Malik R. Guanidine-based β amyloid precursor protein cleavage enzyme 1 (BACE-1) inhibitors for the Alzheimer’s Disease (AD): a review. Bioorg Med Chem. 2022;74:117047. https://doi.org/10.1016/j.bmc.2022.117047.
Anzini M, Chelini A, Mancini A, Cappelli A, Frosini M, Ricci L, et al. Synthesis and biological evaluation of amidine, guanidine, and thiourea derivatives of 2-amino-(6-trifluoromethoxy)benzothiazole as neuroprotective agents potentially useful in brain Diseases. J Med Chem. 2010;53:734–44. https://doi.org/10.1021/jm901375r.
Funding
This work formed part of the first author’s PhD research at the University of Southern Queensland, Australia which has been supported by funding from the Australian Government Research Training Program Scholarship.
Author information
Authors and Affiliations
Contributions
MPM conceptualized, collected data, analyzed, and wrote the first draft of the study. KA, JG, and RAM supervised the study as well as revised and edited the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mosharaf, M.P., Alam, K., Gow, J. et al. Exploration of key drug target proteins highlighting their related regulatory molecules, functional pathways and drug candidates associated with delirium: evidence from meta-data analyses. BMC Geriatr 23, 767 (2023). https://doi.org/10.1186/s12877-023-04457-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12877-023-04457-1