
Abstract
Aim
To discover the novel ATP7B mutations in 103 southern Chinese patients with Wilson disease (WD), and to determine the spectrum and frequency of mutations in the ATP7B gene and genotype–phenotype correlation in a large-scale sample of Chinese WD patients.
Methods
One hundred three WD patients from 101 unrelated families in southern China were enrolled in this study. Genomic DNA was extracted from the peripheral blood. Direct sequencing of all 21 exons within ATP7B was performed. Subsequently, an extensive study of the overall spectrum and frequency of ATP7B mutations and genotype–phenotype correlation was performed in all Chinese patients eligible from the literature, combined with the current southern group.
Results
In 103 patients with WD, we identified 48 different mutations (42 missense mutations, 4 nonsense mutations and 2 frameshifts). Of these, 3 mutations had not been previously reported: c.1510_1511insA, c.2233C>A (p.Leu745Met) and c.3824T>C (p.Leu1275Ser). The c.2333G>T (p.Arg778 Leu) at exon 8, was the most common mutation with an allelic frequency of 18.8%, followed by c.2975C>T (p.Pro992Leu) at exon 13, with an allelic frequency of 13.4%. In the comprehensive study, 233 distinct mutations were identified, including 154 missense mutations, 23 nonsense mutations and 56 frameshifts. Eighty-five variants were identified as novel mutations. The c.2333G>T (p.Arg778 Leu) and c.2975C>T (p.Pro992Leu) were the most common mutations, with allelic frequencies of 28.6% and 13.0%, respectively. Exons 8, 12, 13, 16 and 18 were recognised as hotspot exons. Phenotype–genotype correlation analysis suggested that c.2333G>T (p.Arg778 Leu) was significantly associated with lower levels of serum ceruloplasmin (P = 0.034). c.2975C>T (p.Pro992Leu) was correlated with earlier age of disease onset (P = 0.002). Additionally, we found that the c.3809A>G (p.Asn1270Ser) mutation significantly indicated younger onset age (P = 0.012), and the c.3884C>T (p.Ala1295Val) mutation at exon 18 was significantly associated with hepatic presentation (P = 0.048). Moreover, the patients with mixed presentation displayed the initial WD features at an older onset age than the groups with either liver disease or neurological presentation (P = 0.039, P = 0.015, respectively). No significant difference was observed in the presence of KF rings among the three groups with different clinical manifestations.
Conclusion
In this study, we identified three novel mutations in 103 WD patients from the southern part of China, which could enrich the previously established mutational spectrum of the ATP7B gene. Moreover, we tapped into a large-scale study of a Chinese WD cohort to characterise the overall phenotypic and genotypic spectra and assess the association between genotype and phenotype, which enhances the current knowledge about the population genetics of WD in China.
Similar content being viewed by others


Introduction
Wilson disease (WD), also known as hepatolenticular degeneration (HLD), is an autosomal recessive inherited disorder of copper metabolism, resulting from pathogenic mutations in the ATP7B gene. It is characterised by deficient incorporation of copper into ceruloplasmin and decreased biliary copper excretion, leading to excessive copper accumulation, primarily in the liver, brain and eyes. The toxic deposition of copper in the body results in highly heterogeneous clinical presentations, such as liver impairment, neurological disturbance and/or other derangements [1, 2].
The age of onset ranged from 1 to 72 years. Most of the existing literature regards the worldwide prevalence of WD as approximately 1 in 30,000 to 1 in 50,000, with an estimated carrier rate of 1 in 90 [3]. However, in Korea, where WD is one of the most common inherited metabolic disorders, the carrier frequency and incidence of WD are estimated to be 1 in 88.2 and 1 in 30,778, respectively [4]. In Latvia, the estimated prevalence of WD is 1 in 24,000 cases [5]. Screening of WD in the UK population suggests that the frequency of individuals predicted to carry two mutant pathogenic ATP alleles is 1 in 7026, which is considerably higher than the typically reported prevalence [6]. Ceruloplasmin-based screening for WD in the population of Japan suggested a frequency as high as 1 in 1500 [7].
WD is caused by mutations in the ATP7B gene, discovered in 1993, that encodes a copper transporting P-type ATPase containing 1465 amino acids [8,9,10]. It is located on chromosome 13q14.3 and consists of 21 exons and 20 introns. Genetic disorders of the ATP7B gene disrupt the synthesis and function of the ATP7B protein, and further impair the copper excretion pathway, leading to the abnormal deposition of copper in the body. Currently, there are records of at least 800 distinct disease-causing mutations in the ATP7B gene, characterised by a few hotspot mutations and a wide spectrum of rare mutations, with obvious ethnic and regional differences.
Traditionally, the diagnosis of WD mainly depends upon clinical manifestations and conventional biochemical indicators, including elevated 24-h urinary copper, low serum ceruloplasmin and increased hepatic copper content. However, biochemical tests can be misleading, making WD diagnosis difficult [11, 12]. Hence, molecular detection is warranted for establishing a precise and decisive diagnosis of WD, particularly in asymptomatic patients and siblings of the proband in a WD-affected family. Previous studies on mutations in Chinese WD patients have been based on diverse genetic detection methods with different detection rates. Therefore, a consensus has not yet been reached regarding the spectrum and frequency of mutations in the ATP7B gene in the Chinese WD population. On the other hand, previous studies have failed to identify WD mutations in a significant number of clinically diagnosed cases, resulting in incomplete understanding of the patterns and frequencies of hotspots in the ATP7B gene and controversial correlations between genotypes and phenotypes in the Chinese population with WD.
Here, to obtain the best identification rate and accuracy, we used direct sequencing to detect the WD mutations. This method is considered as the gold standard to identify mutations in molecular genetics [13] and has been documented as having a high detection rate and accuracy [14]. In our study, we first analysed the genotypic profile and determined the novel mutations of 103 cases with WD from the south of China by means of direct sequencing. Subsequently, we conducted a comprehensive literature search for available studies on WD mutations to identify the overall spectrum of ATP7B mutations and the mutation hotspots observed in the Chinese WD population, and to explore the potential correlation between genotype and phenotype.
To the best of our knowledge, this is the first study to undertake a comprehensive literature study to identify the molecular genetic features and correlations with clinical phenotypes in a large-scale sample of Chinese WD patients. Understanding the genotypic pattern of WD in China could pave the way for offering diagnostic mutational analysis of WD in the future. Our genetic investigation of WD patients from the southern part of China could extend the previously established spectrum of ATP7B mutations, and the comprehensive mutation analysis would enhance the current knowledge about the genotypic and phenotypic profiles of WD in China and provides insights into the association between genotype and phenotype in the Chinese population with WD.
Material and methods
Patients
A total of 103 WD patients (66 males and 37 females) from 101 independent families were enrolled in this study, with a mean age at presentation of 18.2 ± 12.9 years. They came from different parts of China (40.9% subjects from Hunan Province, 11.3% from Jiangxi Province, 6.8% from Hubei Province and 4.5% from the northern China). All the patients were identified and diagnosed at the Second Xiangya Hospital in Hunan Province between January 2014 and January 2020. Diagnosis of WD was based on a combination of characteristic clinical symptoms, Kayser–Fleischer (KF) rings, abnormal brain magnetic resonance imaging and biochemical parameters, including low serum ceruloplasmin (< 0.2 g/L), increased urinary copper excretion (> 100 μg/24 h) and high hepatic copper content (> 250 μg/g dry weight). The control group consisted of 37 subjects with neither family history nor clinical features of WD. This study was approved by the local ethics committee, and informed consent was obtained from all recruited subjects or from their parents.
DNA extraction and amplification
The peripheral venous blood was obtained from the WD patients and controls. Genomic DNA was extracted with a Genomic DNA Purification Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Genomic DNA was stored at − 20 °C. ALL 21 exons of the WD gene were amplified by polymerase chain reaction (PCR). The amplification was conducted as follows: pre-denaturation at 94 °C for 5 min; 35 cycles of denaturation at 94 °C for 45 s, annealing at 55 °C for 45 s (except exon 2.2 and exon 17), or annealing at 51 °C for 45 s (to exon 2.2), or annealing at 61 °C for 45 s (to exon17) and extension at 72 °C for 1 min, with a final elongation at 72 °C for 10 min.
DNA sequencing
Direct sequencing of the amplified PCR products was performed to detect the disease-causing mutations and single nucleotide polymorphisms at Sangon Biotech (Shanghai, China), and the sequenced products were compared with published normal sequences deposited in GenBank using a blast search program. Samples showing abnormal results were subjected to bidirectional sequencing.
Systematic review
A comprehensive literature search of PubMed was carried out for articles published from inception until May 2020. Index terms used were Wilson Disease (Title/Abstract) or hepatolenticular degeneration (Title/Abstract). Papers that described the mutations in the WD gene were handpicked by examining online abstracts. Subsequently, further selection was conducted using the full-length publications. Eligibility of the articles was based on the inclusion criteria: (1) observational studies published as original articles that focused on the genetic analysis of WD patients in China; (2) patient cohorts including both paediatric (< 18 years old) and adult (≥ 18 years old) patients; (3) the 21 exons of the ATP7B gene were amplified by PCR; and (4) direct sequencing of the PCR products was performed. We excluded the papers in which the subjects failed to satisfy the foregoing criteria, and which were not written in English. To avoid reporting bias, we manually collected additional relevant studies listed as references of these retrieved articles. This systematic review was conducted in accordance with the PRISMA (preferred reporting items for systematic reviews and meta-analysis) guideline.
A standardised data collection form was utilised to derive the following information: first author, country of origin, year of publication, study design and methods, studied population, gender, age at onset of presentations, WD features (clinical manifestations and biochemical markers), and mutations in ATP7B gene. A systematic analysis for the spectrum of the ATP7B mutations in China was performed on all Chinese WD patients available from qualifying literature, combined with the current southern group. An analysis for the genotype and phenotype correlation in the large-scale Chinese WD cohorts was also performed.
Statistical analysis
Allele and genotype frequencies were calculated by the direct count. Statistical analyses were performed using SPSS for Windows (Version 20.0, SPSS, Inc., Chicago, IL, USA). The distributed analysis of numeric variables was carried out at first. Normally distributed variables were expressed as mean and standard deviation and were compared between groups using the Scheffe test. Variables that were not normally distributed have been presented as median and interquartile range (IQR) and were compared between groups using the Mann–Whitney U test. Frequencies of qualitative variables were compared between groups by the chi-squared (X) test. A P value less than 0.05 was considered statistically significant. Bonferroni correction was applied in case of pairwise comparisons out of larger groups.
Results
Mutation analysis in 103 WD patients
Among the 103 WD patients derived from 101 unrelated families, we have identified 48 distinct mutations, including 42 missense mutations, 4 nonsense mutations and 2 frameshifts, as presented in Table 1. None of these 48 mutations was detected among 68 alleles in healthy individuals. According to the records in the WD mutation database of the University of Alberta (http://www.wilsondisease.med.ualberta.ca/database.asp) and the Human Gene Mutation Database (HGMD) professional (http://www.hgmd.cf.ac.uk), this is the first time that the following mutations have been reported: c.1510_1511insA, c.2233C>A (p.Leu745Met), and c.3824T>C (p.Leu1275Ser). All novel missense variants were tested for the possibility of being pathogenic in nature using PolyPhen-2 software (http://genetics.bwh.harvard.edu/pph2/), with the results indicating that they were significantly more likely to alter protein function, five categories based on ACMG guidelines were used: pathogenic, likely pathogenic, uncertain significance, likely benign, and benign for the variant classification of novel variants using ClinVar database, as shown in Table 1. Moreover, 15 known polymorphisms that do not disrupt ATP7B gene function were detected (data is available in Table 2). Mutation analysis of the ATP7B gene by direct sequencing of 21 exons yielded a mutation detection rate of 80.7% (163/202). There were eight patients having no detectable mutation, indicating that the remaining mutations were possibly located in the intron or the regulator. In the present study, the c.2333G>T (p.Arg778Leu) at exon 8 was the most frequent mutation, with an allelic frequency of 18.8% (38/202), followed by the c.2975C>T (p.Pro992Leu) at exon 13, with an allelic frequency of 13.4% (27/202).
The exons harbouring the highest percentage of mutations were exons 8, 13, 16, 12 and 18. The total mutation detection rate on these five exons was 63.4% (128/202), suggesting that these exons could be important regions for detecting mutations in the southern Chinese WD cohort. The mutations on exons 8 and 13 accounted for 28.8% (n = 47) and 19.6% (n = 32) of the total mutant alleles (n = 163), respectively. The detection rate of other mutations on exons 16, 12 and 18 spread from 14 to 8.0% (Fig. 1).

Distribution of mutations in the ATP7B gene in patients with Wilson disease (WD). The frequency of mutations found in the cohort of 101 WD index cases is given per exon as a percentage of the total mutant alleles
Mutation spectrum of ATP7B in a large-scale sample of Chinese patients
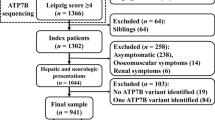
A search of the literature for studies on the overall spectrum of mutations in large-scale sample of Chinese WD patients was conducted. Of the 5,868 studies initially, 5,848 publications were removed due to their irrelevant titles and abstracts. After full-length review of 20 included studies, we further excluded 6 articles that failed to meet the inclusion criteria. Finally, 14 eligible articles with 1201 WD probands were included, and an additional 101 probands from the southern part of China in the present study were also included, for a grand total of 1302 index patients with WD in the systemic analysis. In total, 233 different mutations in the coding region of the ATP7B gene were detected in our patient pool, including 154 missenses, 23 nonsenses and 56 insertions or deletions. Eighty-five variants were identified as novel mutations in the WD databases mentioned above. The computational predictive analysis of the missense variants by PolyPhen-2 was also shown in Table 1. Most of the missense substitutions showed a significant effect on the protein. All mutations accounted for 87.0% (n = 2265) of the alleles studied (n = 2604), with c.2333G>T (p.Arg778Leu) and c.2975C>T (p.Pro992Leu) being the two most common mutations at a frequency of 28.6% and 13.0%, respectively. Mutations were distributed in all exons except exon 1. We found that exons 8, 13, 12 and 16 were the hotspot exons in this large WD pool, accounting for 64.1% (1668/2604) of studied alleles. The mutations accumulated on exons 8, 13, 12 and 16 showed higher prevalence, accounting for 38.0%, 17.3%, 10.9% and 7.4% of the mutant chromosomes, respectively, as depicted in Fig. 2.

Distribution and frequency of mutations in the ATP7B gene in 1302 patients with Wilson disease in the Chinese population
It was interesting to note that the prevalent mutation c.2333G>T (p.Arg778Leu) was almost exclusively linked with c.2310C>G (p.Leu770Leu) polymorphism. C.2310C>G (p.Leu770Leu) polymorphism was rare in the normal population but appeared frequently in the WD cohort, suggesting that, to some extent, this linkage may impact the expression of the ATP7B protein.
Characterisation of clinical phenotype
Screening patients from the 14 eligible reports [14,15,16,17,18,19,20,21,22,23,24,25,26,27] identified 108 patients with clear WD features. We enrolled 196 patients with WD in the final analysis, including the current 88 patients with detailed data from southern China. Of these, 50.5% (99/196) patients had a primary hepatic manifestation, 18.9% (37/196) showed a primary neurological manifestation and 13.8% (27/196) had combined hepatic and neurological manifestations. Thirty-three of the 196 patients (16.8%) presented with no symptoms. The mean age at symptom onset was 19.3 years (from 1 to 62 years). The median ceruloplasmin level was 82.2 mg/L (range 20–962 mg/L).
We observed that the patients with mixed manifestation were significantly older at symptom onset than patients with hepatic symptoms (24.4 V 19.4 years of age, P = 0.039) and patients with neurological symptoms (24.4 V 17.9 years of age, P = 0.015). There is no significant difference in terms of onset age between patients with hepatic phenotype and patients with neurological presentation (P > 0.05). In addition, as shown in Fig. 3A, the patients with typically clinical manifestations displayed later age of onset than the clinically asymptomatic patients, which was a statistically significant difference (19.8 V 12.9 years of age, P = 0.000). No significant difference was observed in the presence of KF rings among the three clinical subtypes with different manifestations (Fig. 3B).

Correlations of four clinical subtypes and A onset age and B the presence of cornea Kayser–Fleischer (KF) ring. A The age of onset was older in the M group than that in the H (P = 0.039) and N (P = 0.015) groups. The onset age was similar between the H and N groups (P > 0.05). The presymptomatic group displayed younger onset age than the H, N and M groups (P = 0.000). B The presence of KF rings was not significantly different among the three groups with different clinical presentations (P > 0.05). H hepatic subtype, N neurological subtype, M mixed subtype, Pre presymptomatic subtype
Correlation between genotype and phenotype
To describe the picture of correlation between genotype and phenotype based on the cohort investigation, we initially studied the rarely reported association between the exons and clinical subtypes. First, we examined the hotspot exons in the available 196-patient WD cohort. The results showed that exons 8, 13 and 16 harboured the highest percentage of mutations, consistent with the results described in the large WD patient pool in the comprehensive analysis. Secondly, in different clinical subtypes, we sorted 21 exons in order of mutation frequencies to gain more insights into the most frequent exons in different types of clinical presentations. Notably, in the mixed presentation group, the second most prevalent exon was exon 11, found in 11.5% of mutant alleles, which was much higher than that in the other two groups. Hence, we assumed that the mutations in exon 11 might play an important role in the development of combined presentation. In the primary hepatic group, exon 18 was the third most mutant exon, contributing 10.4% of mutant chromosomes. The possible association between the mutations in exon 18 and the hepatic manifestation is discussed below. In this course, we observed that c.2621C>T (p.Ala874Val), the most prevalent mutation in exon 11, frequently occurred with c.2333 G>T (p.Arg778Leu) except in single heterozygotes. Another observation was that c.3884C>T (p.Ala1295Val), the most prevalent mutation in exon 18, only mutated in patients with hepatic symptoms. The difference between the c.3884C>T (p.Ala1295Val) patients with non-c.3884C>T (p.Ala1295Val) patients in three clinical groups was statistically significant (P = 0.048), as shown in Fig. 4A. This indicated that the c.3884C>T (p.Ala1295Val) mutation in exon 18 was significantly associated with hepatic symptoms.

A Correlation of 3884C>T (Ala1295Val) and clinical manifestations; B correlation of 2333G>T (Arg778 Leu) and serum ceruloplasmin level; C correlation of 2975C>T (Pro992Leu) and the onset age; D correlation of 3809 A>G (Asn1270Ser) and the onset age. Homo homozygotes for the mutation, Hetero heterozygotes for the mutation, H hepatic manifestation, N neurological manifestation, M mixed manifestation
Next, we studied the correlation between specific mutations and the phenotypes. We examined the most prevalent mutations in the available 196-patient WD cohort. The results showed that the two most common mutations were c.2333G>T (p.Arg778Leu) and c.2975C>T (p.Pro992Leu), accounting for 21.4% (84/392) and 12.5% (49/392) of the alleles, in good agreement with the results demonstrated in the comprehensive analysis. With regard to the most frequent mutation, c.2333 G>T (p.Arg778Leu), we observed that patients carrying the c.2333 G>T (p.Arg778Leu) mutation had a lower serum ceruloplasmin levels than patients with other mutations at both alleles. When comparing c.2333 G>T (p.Arg778Leu) homozygous (39.3 ± 23.5 mg/L) or heterozygous patients (67.7 ± 48.1 mg/L) with non-c.2333 G>T (p.Arg778Leu) patients (79.7 ± 47.1 mg/L), we found significant differences (P = 0.018, P = 0.049, respectively). However, the difference between c.2333G>T (p.Arg778Leu) homozygous and heterozygous patients was not significant, as shown in Fig. 4B. With regard to the second most common mutation, c.2975 C>T (p.Pro992Leu), we found that the patients with c.2975 C>T (p.Pro992Leu) mutation often exhibited symptoms earlier than the patients without c.2975 C>T (p.Pro992Leu)mutation at both chromosomes (Fig. 4C). The difference in the age of onset between c.2975 C>T (p.Pro992Leu) homozygous (9.7 ± 4.2 years of age) and non-c.2975 C>T (p.Pro992Leu) patients (20.5 ± 12.2 years of age) was significant (P = 0.01), and the difference between c.2975 C>T (p.Pro992Leu) heterozygous (15.7 ± 11.6 years of age) and non-c.2975 C>T (p.Pro992Leu) patients was also significantly different (P = 0.017). No significant difference was observed in the age of onset between c.2975 C>T (p.Pro992Leu) homozygous and heterozygous patients (P > 0.05). We also found a dramatic association between the c.3809 A>G (p.Asn1270Ser) mutation and the disease onset age. Statistics showed that the patients with c.3809 A>G (p.Asn1270Ser) mutation had an earlier age of onset (10.8 ± 7.4 years of age) than the non-c.3809 A>G (p.Asn1270Ser) patients (19.3 ± 12.0 years of age) (P = 0.012, Fig. 4D).
It should be noted that 82.2% of patients carried at least two mutations at both alleles, while 17.8% patients only carried one mutation at two chromosomes. We did not find a significant correlation between the different forms of mutation (homozygous V heterozygous mutations, combined mutations V single heterozygous mutations) and the several clinical indices in terms of the age of onset, clinical manifestations, ceruloplasmin level and the presence of KF rings.
Discussion
In the present study, we explored the mutations in the ATP7B gene in 101 WD probands from southern China. Forty-eight mutations were found, including 3 novel variants. These novel variants were not found in the control chromosomes. Substituted amino acids with a PolyPhen-2 score close to 1.000 could be predicted to be potentially damaging. However, insertion, deletion and premature stop mutations failed to yield acceptable results from PolyPhen-2 analysis. We regarded the insertion mutation (c.1510–1511 insA) as a clearly pathogenic mutation since it caused a frameshift leading to a premature stop codon. According to the PolyPhen-2 score, the other two variants, c.2233 C>A (p.Leu745Met), c.3824T>C (p.Leu1275Ser)) were both predicted to affect protein function. A mutation detection rate of 80.7% was achieved in the southern cohort, but 19.3% of alleles remained unidentified. One study showed that the rate of mutation detection in this study was 83.8% (67/80) of alleles on direct sequencing of the PCR products of all exons of the ATP7B gene in the 40 unrelated Chinese patients with WD [18]. Rui Hua performed mutational analysis of 68 WD patients from China and found that the rate of mutation detection was up to 97.1% [19]. Failure to detect the remaining mutations may be explained by some objective factors, such as the primers, the PCR procedure and/or the sequence alignment. In our recent studies, the detection rate of direct sequencing could reach as high as 95% with the newly designed PCR primers and the improved amplification requirements. Another reason may be due to the presence of mutations outside the open reading frame of the gene, i.e., in the promoter, introns, the presence of gene rearrangements or possible mutations in other copper-transport chaperone gene. Anna Kluska proved that rare allelic variants in ESD and IN080 increased and decreased the chances for the neurologic phenotype, respectively, while rare variants in APOE and MBD6 decreased the possibilities of WD early manifestation [28]. It was reported that the AmpliSeq Exome kit usually underestimated the insertions and deletions in exome enrichment products [28].
C.2333G>T (p.Arg778Leu) was the most frequent mutation in our study and was also described as the most common mutation in China [16,17,18,19,20,21, 23, 24], accounting for 18.8% of alleles studied here. The second most common mutation was c.2975 C>T (p.Pro992Leu) among the WD patients, with an allelic frequency of 13.4%, consistent with the frequency previously reported in China [17, 19, 20, 23]. However, Hong et al. [24] suggested that c.3443T>C (p.Ile1148Thr) was the second most common mutation instead of c.2975 C>T (p.Pro992Leu) in their cohort study of 103 Chinese WD patients. An earlier study of 114 WD patients from northern China demonstrated that c.2621 C>T (p.Ala874Val) was the second hot-spot mutation, followed by c.2975 C>T (p.Pro992Leu), at an allelic frequency of 6.1% [29]. We speculate that different gene-level tests or a limited number of patients is largely responsible for the differential conclusions. A large-scale or prospective study, based on the same detection standard, is imperative.
In our current study, exon 8 was the most frequent mutational site, found in 28.8% of mutant alleles, followed by exon 13 in 19.6% and exon 16 in 14.1%, indicating that these three exons could be important regions for detecting mutations.
We conducted a comprehensive analysis of the spectrum and frequency of ATP7B mutations in a large-scale sample of Chinese WD patients from more than 30 provinces, autonomous regions and municipalities of China. A total of 233 distinct mutations were detected, of which 85 were novel. The computational predictive analysis software PolyPhen-2 interpreted most of the novel missense variants as disease-causing mutations, with the exception of one benign variant (c.2261A>G, (p.Glu754Gly)). It cannot be ruled out that the silent mutation interpreted as benign could affect protein function.
The most prevalent mutation in the 1302 WD patients pool was c.2333G>T (p.Arg778Leu), in exon 8, with an allelic frequency of 28.6%. The c.2333G>T (p.Arg778Leu) mutation is frequently found in reports of Asian patients, with an allele frequency of 12 to 50% [14, 16,17,18,19,20, 30]. In contrast, the c.3207 C>A (p.His1069Gln) mutation, the most common mutation in European and North American populations, accounting for 30 to 70% of the alleles studied [30], was not detected in any Chinese patients. The next most frequent mutation in this large cohort of Chinese patients was c.2975 C>T (p.Pro992Leu), with an allelic frequency of 13.0%. To our knowledge, the highest frequency of c.2975 C>T (p.Pro992Leu) described so far was 27% [14].
All exons except exon 1 were affected. Notably, exons 8, 13, 12 and 16 were the hot-spot exons identified in the large WD population, accounting for 73.6% of mutant alleles, consistent with previous results that 60.5 to 74% of mutations were located on the above hot-spot exons [31].
The spectrum of WD mutations in the large cohort of Chinese patients consisted of a small number of relatively frequent mutations and a greater number of rare mutations. This further indicated a high degree of mutational heterogeneity, in agreement with previously published findings [32, 33]. Moreover, we found that many mutations were located a short distance away, in line with the preliminary results [34]. Additionally, 64.3% patients were found to stay in a compound heterozygotic state, compared with 13.8% patients in a homozygotic state and 17.4% patients in a single heterozygotic state, which can be explained by the low percentage of consanguinity in our investigated population. No significant difference in phenotypic profiles were found when comparing homozygous or combined heterozygous patients with the patients who had only one mutation at two alleles. We suspected that, to the patients with a single mutation, the remaining unidentified mutations would probably be located in non-coding regions of the ATP7B gene. Other mutational mechanisms should also be taken into consideration.
One polymorphism with substitution of leucine with leucine at codon 770 in the transmembrane region of ATP7B has been found to be linked with the c.2333 G>T (p.Arg778Leu) mutation. Perhaps, the coexistence of the c.2333G>T (p.Arg778Leu) mutation and the c.2310C>G (p.Arg778Leu) polymorphism would have a special effect on the ATP7B protein. Further investigation of the functional implications of both is needed.
A well-defined landscape of the genotype–phenotype correlation will promote the development of clinical studies. However, most of previous studies devoted to genotype–phenotype association have addressed rare or conflicting conclusions [35, 36]. The His1069Gln mutation is most common on Western populations. Genotype–phenotype correlation studies indicated that the His1069Gln mutation was associated more frequently with neurological phenotype [37]. However, the studies in 126 Bulgarian patients presenting a His1069Gln allele frequency in 78% of cases indicated a correlation between that variant and hepatic presentation [38]. Tarnacka et al. reported on 148 Polish patients with a high p.His1069Gln frequency and did not find any association between genotype and phenotype [39]. The studies in Chinese patients showed an association between homozygous p.Arg778Leu and neurologic phenotype [20]. One reported the mutation p.Pro992Leu contributed to early onset age in WD patients, but they did not report any association between p.Arg778Leu mutation and clinical presentation [19]. Verification of this requires a cohort study. Our study significantly described a systemic and quantitative analysis of the genotype–phenotype correlation in a large cohort of Chinese patients with WD.
In the demonstration of hot-spot exons in different types of clinical presentations, we identified that exon 11 was ranked as the second most mutational exon in the mixed presentation. The difference in the proportion of patients with mutations in exon 11 between the hepatic and the mixed group was significant (P = 0.046), while the difference between the neurological and the mixed group was not significant. We could not reach the correlation between the mutations in exon 11 and the mixed manifestation. C.2621 C>T (p.Ala874Val), the predominant mutation in exon 11, frequently presented with c.2333 G>T (p.Arg778Leu) substitution. Krishna et al. considered that the hydrophobicity and conformational stability of the hydrophobic domains, such as transmembrane domains, may be altered due to the valine amino acid [40]. We speculated that the transmembrane domain region of ATP7B with valine at the 874 domain region and with leucine at 778 could probably destabilise the formation or influence the expression of protein. Functional studies of mutations are required for the validation of our speculation. Another finding in the analysis of exon hotspots in different clinical presentations was that exon 18 ranked as the third exon with the most mutations in the hepatic presentation group, with a higher mutation frequency (10.38%) than that in the other two groups (6.25% and 3.85% in the neurological and mixed presentation, respectively). This is probably attributable to the potential association between the mutations in exon 18 and the hepatic involvement. Fortunately, we identified that c.3884 C>T (p.Ala1295Val), one kind of the mutations in exon 18, only mutated in the patients with hepatic manifestation. Statistical analysis revealed that there was a significant association between the c.3884 C>T (p.Ala1295Val) mutation and the hepatic phenotype, which was consistent with previous observations that mutations in the conserved ATP hinge region were associated with liver disease without neurological presentation [41], and when the mutation affected the ATP hinge, it resulted in hepatic failure [42].
Furthermore, we found a statistically significant correlation between the c.2333 G>T (p.Arg778Leu) mutation and lower serum ceruloplasmin levels. The difference in the serum ceruloplasmin level between c.2333 G>T (p.Arg778Leu) homozygous or heterozygous patients and non-c.2333 G>T (p.Arg778Leu) patients was significant. A recent study in a large cohort of Chinese WD patients [43] showed that c.2333 G>T (p.Arg778Leu) was related to lower levels of ceruloplasmin as well. That study also suggested that c.2333 G>T (p.Arg778Leu) was related to younger onset age. However, in our study, we did not find the significant difference between c.2333 G>T (p.Arg778Leu) and the onset age, and we did not find a considerable difference between c.2333 G>T (p.Arg778Leu) and the hepatic manifestation either, as previously reported by Liu et al. [44]. Significant difference in the age of onset was observed between c.2975 C>T (p.Pro992Leu) homozygous or combined heterozygous patients and non-2975 C>T (p.Pro992Leu) patients. Collectively, our finding revealed that the patients with c.2975 C>T (p.Pro992Leu) mutation often presented with WD profiles at an earlier age, usually before 13.7 years old, than the patients with other mutations, while Hua et al. [19] described that the patients with c.2975C>T (p.Pro992Leu) often presented WD features before 12 years old. We also found a remarkable association between c.3809 A>G (p.Asn1270Ser) and the disease onset age. Statistical findings showed that the patients with c.3809 A>G (p.Asn1270Ser) mutation usually manifested the WD features before 10.8 years old, much earlier than the patients with other mutations at two chromosomes.
We also observed a visible correlation between the onset age and the characteristic clinical manifestations. Our findings showed that the patients with mixed manifestation had a later age of onset than the groups with either liver disease or neurological phenotype. However, the two latter onset ages were not significantly different from one another, inconsistent with the understanding that patients having predominantly neuropsychiatric symptoms usually manifest symptoms later than patients with hepatic presentation [45,46,47]. Our results showed that the liver and brain could be affected by WD simultaneously.
In addition, we discovered interesting clinical differences between the symptomatic group and the asymptomatic group in terms of the presence of KF rings. KF rings of symptomatic patients were found to be significantly higher than KF rings of asymptomatic cases, consistent with the earlier findings [48]. We also found that the patients who had KF rings were significantly older at symptom onset than the cohort without KF rings, which is in line with the finding identified in our study that the patients with typical clinical manifestations significantly displayed later age of onset than the patients who were clinically asymptomatic. In summary, the cohort with clinical symptoms presented with a later age of onset and higher prevalence of KF rings than the asymptomatic cohort. One report claimed that patients with purely neurological symptoms were susceptible to KF rings [49]. Our current study did not show any clear difference in the presence of KF rings among the three groups with different clinical manifestations.
In conclusion, we characterised a complete genotypic and phenotypic profile of Chinese patients with WD. The three novel mutations identified in the southern Chinese WD patients could considerably extend the previously established spectrum of the ATP7B mutations. Comprehensive mutation analysis will enhance the current knowledge of WD genetics in China. The findings of correlation between specific mutations and clinical features, as well as the age of onset and several clinical profiles provides new insights into the relationships between genotype and phenotype. Additional large studies are required for validation of our conclusions.
Availability of data and materials
All data generated or analysed during this study are included in this published article, except the sequencing data. All clean sequence data were deposited in the NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/Traces/sra/) under the accession numbers SRR9969677-SRR9969698.
References
Mulligan C, Bronstein JM. Wilson disease: an overview and approach to management. Neurol Clin. 2020;38(2):417–32. https://doi.org/10.1016/j.ncl.2020.01.005.
Chaudhry HS, Anilkumar AC. Wilson disease. StatPearls Publishing; 2021. [PMID: 28723019, Bookshelf ID: NBK441990].
Sandahl TD, Laursen TL, Munk DE, Vilstrup H, Weiss KH, Ott P. The prevalence of Wilson’s disease: an update. Hepatology. 2020;71(2):722–32. https://doi.org/10.1002/hep.30911.
Kim GH, Yang JY, Park JY, Lee JJ, Kim JH, Yoo HW. Estimation of Wilson’s disease incidence and carrier frequency in the Korean population by screening ATP7B major mutations in newborn filter papers using the SYBR green intercalator method based on the amplification refractory mutation system. Genet Test. 2008;12(3):395–9. https://doi.org/10.1089/gte.2008.0016.
Zarina A, Tolmane I, Kreile M, Chernushenko A, Cernevska G, Pukite I, Micule I, Krumina Z, Krumina A, Rozentale B, Piekuse L. Genetic variation spectrum in ATP7B gene identified in Latvian patients with Wilson disease. Mol Genet Genomic Med. 2017;5(4):405–9. https://doi.org/10.1002/mgg3.297.
Coffey AJ, Durkie M, Hague S, McLay K, Emmerson J, Lo C, Klaffke S, Joyce CJ, Dhawan A, Hadzic N, Mieli-Vergani G, Kirk R, Elizabeth Allen K, Nicholl D, Wong S, Griffiths W, Smithson S, Giffin N, Taha A, Connolly S, Gillett GT, Tanner S, Bonham J, Sharrack B, Palotie A, Rattray M, Dalton A, Bandmann O. A genetic study of Wilson’s disease in the United Kingdom. Brain. 2013;136(Pt 5):1476–87. https://doi.org/10.1093/brain/awt035.
Ohura T, Abukawa D, Shiraishi H, Yamaguchi A, Arashima S, Hiyamuta S, Tada K, Iinuma K. Pilot study of screening for Wilson disease using dried blood spots obtained from children seen at outpatient clinics. J Inherit Metab Dis. 1999;22(1):74–80.
Petrukhin K, Fischer SG, Pirastu M, Tanzi RE, Chernov I, Devoto M, Brzustowicz LM, Cayanis E, Vitale E, Russo JJ. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet. 1993;5(4):338–43. https://doi.org/10.1038/ng1293-338.
Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5(4):327–37. https://doi.org/10.1038/ng1293-327.
Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, Romano DM, Parano E, Pavone L, Brzustowicz LM. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5(4):344–50. https://doi.org/10.1038/ng1293-344.
Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089–111. https://doi.org/10.1002/hep.22261.
Medici V, Rossaro L, Sturniolo GC. Wilson disease—a practical approach to diagnosis, treatment and follow-up. Dig Liver Dis. 2007;39(7):601–9. https://doi.org/10.1016/j.dld.2006.12.095.
Poon KS, Tan KM, Koay ES. Targeted next-generation sequencing of the ATP7B gene for molecular diagnosis of Wilson disease. Clin Biochem. 2016;49(1–2):166–71. https://doi.org/10.1016/j.clinbiochem.2015.10.003.
Ye S, Gong L, Shui QX, Zhou LF. Wilson disease: identification of two novel mutations and clinical correlation in Eastern Chinese patients. World J Gastroenterol. 2007;13(38):5147–50.
Dong Y, Ni W, Chen WJ, Wan B, Zhao GX, Shi ZQ, Zhang Y, Wang N, Yu L, Xu JF, Wu ZY. Spectrum and classification of ATP7B variants in a large cohort of chinese patients with Wilson’s disease guides genetic diagnosis. Theranostics. 2016;6(5):638–49. https://doi.org/10.7150/thno.14596.
Geng J, Wang J, Yao RE, Liu XQ, Fu QH. Identification of one novel and nine recurrent mutations of the ATP7B gene in 11 children with Wilson disease. World J Pediatr. 2013;9(2):158–62. https://doi.org/10.1007/s12519-012-0388-7.
Gu S, Yang H, Qi Y, Deng X, Zhang L, Guo Y, Huang Q, Li J, Shi X, Song Z, Deng H. Novel ATPase Cu (2+) transporting beta polypeptide mutations in Chinese families with Wilson’s disease. PLoS ONE. 2013;8(7):e66526. https://doi.org/10.1371/journal.pone.0066526.
Gu YH, Kodama H, Du SL, Gu QJ, Sun HJ, Ushijima H. Mutation spectrum and polymorphisms in ATP7B identified on direct sequencing of all exons in Chinese Han and Hui ethnic patients with Wilson’s disease. Clin Genet. 2003;64(6):479–84.
Hua R, Hua F, Jiao Y, Pan Y, Yang X, Peng S, Niu J. Mutational analysis of ATP7B in Chinese Wilson disease patients. Am J Transl Res. 2016;8(6):2851–61.
Li XH, Lu Y, Ling Y, Fu QC, Xu J, Zang GQ, Zhou F, De-Min Y, Han Y, Zhang DH, Gong QM, Lu ZM, Kong XF, Wang JS, Zhang XX. Clinical and molecular characterization of Wilson’s disease in China: identification of 14 novel mutations. BMC Med Genet. 2011;12:6. https://doi.org/10.1186/1471-2350-12-6.
Liu Y, Zhou H, Guo H, Bai Y. Genetic and clinical analysis in a cohort of patients with Wilson’s disease in Southwestern China. Arch Med Res. 2015;46(2):164–9. https://doi.org/10.1016/j.arcmed.2015.02.001.
Lu CX, Lin Q, Huang WQ, Tzeng CM. New mutations and polymorphisms of the ATP7B gene in sporadic Wilson disease. Eur J Med Genet. 2014;57(9):498–502. https://doi.org/10.1016/j.ejmg.2014.04.016.
Wan L, Tsai CH, Hsu CM, Huang CC, Yang CC, Liao CC, Wu CC, Hsu YA, Lee CC, Liu SC, Lin WD, Tsai FJ. Mutation analysis and characterization of alternative splice variants of the Wilson disease gene ATP7B. Hepatology. 2010;52(5):1662–70. https://doi.org/10.1002/hep.23865.
Wei Z, Huang Y, Liu A, Diao S, Yu Q, Peng Z, Hong M. Mutational characterization of ATP7B gene in 103 Wilson’s disease patients from Southern China: identification of three novel mutations. NeuroReport. 2014;25(14):1075–80. https://doi.org/10.1097/WNR.0000000000000216.
Zhang DF, Teng JF. Direct sequencing of mutations in the copper-transporting P-type adenosine triphosphate (ATP7B) gene for diagnosis and pathogenesis of Wilson’s disease. Genet Mol Res. 2016;15(3):gmr.15038746. https://doi.org/10.4238/gmr.15038746.
Zhu HW, Tao ZB, Su G, Jin QY, Zhao LT, Zhu JR, Yan J, Yu TY, Ding JX, Li YM. Identification of two novel mutations in the ATP7B gene that cause Wilson’s disease. World J Pediatr. 2017;13(4):387–91. https://doi.org/10.1007/s12519-017-0055-0.
Chen L, Li X, Zheng Z, Lu X, Lin M, Pan C, Liu J. A novel ATP7B gene mutation in a liver failure patient with normal ceruloplasmin and low serum alkaline phosphatase. Gene. 2014;538(1):204–6. https://doi.org/10.1016/j.gene.2013.10.044.
Kluska A, Kulecka M, Litwin T, Dziezyc K, Balabas A, Piatkowska M, Paziewska A, Dabrowska M, Mikula M, Kaminska D, Wiernicka A, Socha P, Czlonkowska A, Ostrowski J. Whole-exome sequencing identifies novel pathogenic variants across the ATP7B gene and some modifiers of Wilson’s disease phenotype. Liver Int. 2019;39(1):177–86. https://doi.org/10.1111/liv.13967.
Li K, Zhang WM, Lin S, Wen L, Wang ZF, Xie D, Wei M, Qiu ZQ, Dai Y, Lin MC, Kung HF, Yao FX. Mutational analysis of ATP7B in north Chinese patients with Wilson disease. J Hum Genet. 2013;58(2):67–72. https://doi.org/10.1038/jhg.2012.134.
Ferenci P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet. 2006;120(2):151–9. https://doi.org/10.1007/s00439-006-0202-5.
Wu ZY, Wang N, Lin MT, Fang L, Murong SX, Yu L. Mutation analysis and the correlation between genotype and phenotype of Arg778Leu mutation in Chinese patients with Wilson disease. Arch Neurol. 2001;58(6):971–6.
Al Jumah M, Majumdar R, Al Rajeh S, Awada A, Al Zaben A, Al Traif I, Al Jumah AR, Rehana ZA. Clinical and genetic study of 56 Saudi Wilson disease patients: identification of Saudi-specific mutations. Eur J Neurol. 2004;11(2):121–4.
Vrabelova S, Letocha O, Borsky M, Kozak L. Mutation analysis of the ATP7B gene and genotype/phenotype correlation in 227 patients with Wilson disease. Mol Genet Metab. 2005;86(1–2):277–85. https://doi.org/10.1016/j.ymgme.2005.05.004.
Haas R, Gutierrez-Rivero B, Knoche J, Boker K, Manns MP, Schmidt HH. Mutation analysis in patients with Wilson disease: identification of 4 novel mutations. Mutation in brief no. 250. Hum Mutat. 1999;14(1):88. https://doi.org/10.1002/(SICI)1098-1004(1999)14:1%3c88::AID-HUMU15%3e3.0.CO;2-H.
Peter F, Wolfgang S, et al. Age and sex but not ATP7B genotype effectively influences the clinical phenotype of Wilson disease. Hepatology. 2019;69(4):1464–76. https://doi.org/10.1002/hep.30280.
Medici V, LaSalle JM. Genetics and epigenetic factors of Wilson disease. Ann Transl Med. 2019;7(Suppl 2):S58. https://doi.org/10.21037/atm.2019.01.67.
Caca K, Ferenci P, Kühn HJ, Polli C, Willgerodt H, Kunath B, Hermann W, Mössner J, Berr F. High prevalence of the H1069Q mutation in East German patients with Wilson disease: rapid detection of mutations by limited sequencing and phenotype–genotype analysis. J Hepatol. 2001;35(5):575–81. https://doi.org/10.1016/s0168-8278(01)00219-7.
Mihaylova V, Todorov T, Jelev H, Kotsev I, Angelova L, Kosseva O, Georgiev G, Ganeva R, Cherninkova S, Tankova L, Savov A, Tournev I. Neurological symptoms, genotype–phenotype correlations and ethnic-specific differences in Bulgarian patients with Wilson disease. Neurologist. 2012;18(4):184–9. https://doi.org/10.1097/NRL0bcf3b7.
Tarnacka B, Gromadzka G, Rodo M, Mierzejewski P, Czloonkowska A. Frequency of His1069Gln and Gly1267Lys mutations in Polish Wilson’s disease population. Eur J Neurol. 2000;7(5):495–8. https://doi.org/10.1046/1468-1331.2000.t01-1-00112.x.
Krishna BM, Chaudhary S, Panda AK, Mishra DR, Mishra SK. Her2 (Ile)655(Val) polymorphism and its association with breast cancer risk: an updated meta-analysis of case-control studies. Sci Rep. 2018;8(1):7427. https://doi.org/10.1038/s41598-018-25769-y.
Barada K, El-Atrache M, El Hajj II, Rida K, El-Hajjar J, Mahfoud Z, Usta J. Homozygous mutations in the conserved ATP hinge region of the Wilson disease gene: association with liver disease. J Clin Gastroenterol. 2010;44(6):432–9. https://doi.org/10.1097/MCG.0b013e3181ce5138.
Abdelghaffar TY, Elsayed SM, Elsobky E, Bochow B, Buttner J, Schmidt H. Mutational analysis of ATP7B gene in Egyptian children with Wilson disease: 12 novel mutations. J Hum Genet. 2008;53(8):681–7. https://doi.org/10.1007/s10038-008-0298-7.
Cheng N, Wang H, Wu W, Yang R, Liu L, Han Y, Guo L, Hu J, Xu L, Zhao J, Han Y, Liu Q, Li K, Wang X, Chen W. Spectrum of ATP7B mutations and genotype–phenotype correlation in large-scale Chinese patients with Wilson Disease. Clin Genet. 2017;92(1):69–79. https://doi.org/10.1111/cge.12951.
Liu XQ, Zhang YF, Liu TT, Hsiao KJ, Zhang JM, Gu XF, Bao KR, Yu LH, Wang MX. Correlation of ATP7B genotype with phenotype in Chinese patients with Wilson disease. World J Gastroenterol. 2004;10(4):590–3.
Aggarwal A, Chandhok G, Todorov T, Parekh S, Tilve S, Zibert A, Bhatt M, Schmidt HH. Wilson disease mutation pattern with genotype–phenotype correlations from Western India: confirmation of p.C271* as a common Indian mutation and identification of 14 novel mutations. Ann Hum Genet. 2013;77(4):299–307. https://doi.org/10.1111/ahg.12024.
Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study. Gut. 2007;56(1):115–20. https://doi.org/10.1136/gut.2005.087262.
Kumar S, Thapa B, Kaur G, Prasad R. Analysis of most common mutations R778G, R778L, R778W, I1102T and H1069Q in Indian Wilson disease patients: correlation between genotype/phenotype/copper ATPase activity. Mol Cell Biochem. 2007;294(1–2):1–10. https://doi.org/10.1007/s11010-005-9028-z.
Rodman R, Burnstine M, Esmaeli B, Sugar A, Martonyi C, Johnson V, Brewer G. Wilson’s disease: presymptomatic patients and Kayser–Fleischer rings. Ophthalmic Genet. 1997;18(2):79–85.
Seo GH, Kim YM, Oh SH, Chung SJ, Choi IH, Kim GH, Yum MS, Choi JH, Kim KM, Ko TS, Lee BH. Biochemical and molecular characterisation of neurological Wilson disease. J Med Genet. 2018. https://doi.org/10.1136/jmedgenet-2017-105214.
Acknowledgements
We thank all the patients who agreed to participate in this study.
Funding
This study was supported by the Hunan Province Development and Reform Commission Foundation (KZ, Luo N0. 2016-65), the Key R&D Program of Hunan province (No. 2020SK30291, No. 2020SK2083), the National Key R&D Program of China (No. 2019YFE0190800), and the National Natural Science Foundation of China (No. 81500455, No. 81974079). The funders had no influence on the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
XY and KL designed the experiments. ML developed the method and executed the experiments. JM and WW performed Statistical analysis. ML wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Central South University Medical College (Reference number: S073(2013)). The study was conducted in accordance with relevant guidelines and regulations. All methods were performed in accordance with the Declaration of Helsinki. Informed consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, M., Ma, J., Wang, W. et al. Mutation analysis of the ATP7B gene and genotype–phenotype correlation in Chinese patients with Wilson disease. BMC Gastroenterol 21, 339 (2021). https://doi.org/10.1186/s12876-021-01911-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-021-01911-5