
Abstract
Background
Immunoglobulin G4 (IgG4) associated autoimmune hepatitis (AIH) has been recognized as a type of autoimmune disease that responds to corticosteroid. The diagnosis is based on elevation of the serum IgG4 level, abundance of IgG4 enhanced plasma cell infiltration in the portal region of the liver, and satisfaction of the criteria for “definite AIH” under the revised International Autoimmune Hepatitis Group (IAIHG) scoring system. However, the clinical course of the disease is unclear.
Case presentation
A 65-year-old man with jaundice and peripheral blood eosinophilia. His IAIHG and simplified score was compatible with definite AIH and his IgG4 level was elevated. Magnetic resonance imaging did not reveal abnormalities in the hepatobiliary system or pancreas. A liver biopsy revealed interface hepatitis with IgG4 positive plasma cell infiltration in the portal region, without evidence of bile duct injury. He responded to 4-week period of induction prednisolone therapy and had no recurring symptoms under maintenance therapy of 5 mg prednisolone during the 3-year follow up.
Conclusions
This was a rare case that demonstrated an association between IgG4 associated AIH and the presence of peripheral blood eosinophilia.
Similar content being viewed by others


Background
Autoimmune hepatitis (AIH) is an inflammatory liver disease characterized by chronic inflammation of the liver, positivity for autoantibodies, increased immunoglobulin level, and histological evidence of interface hepatitis and lymphoplasmacytic infiltration [1]. In contrast, immunoglobulin G4 (IgG4)-related disease is a chronic, relapsing, systemic, fibro-inflammatory syndrome of presumed autoimmune etiology with high blood levels of IgG4, IgG, and IgE [2]. The association between high serum IgG4 level and high peripheral eosinophil count has been proved; peripheral blood and tissue eosinophilia have been observed in some IgG4-related-disease patients [3]. Recently, AIH with an elevated serum IgG4 concentration and an abundance of IgG4-positive plasma cell infiltration in the liver was proposed to be termed “IgG4 associated AIH” and regarded as a subtype of IgG4-related disease [4]. However, there are few reported cases of IgG4 associated AIH (IgG4-AIH), and the clinical course of this disease remains poorly understood. Here, we report an interesting case of a patient with peripheral blood eosinophilia who was diagnosed with IgG4-AIH and review the literature on this topic.
Case presentation
A 64-year-old man presented with a 2-month history of progressive, painless jaundice, loss of appetite, and a recent weight loss of 6-kg. He reported a history of uninvestigated, self-limited jaundice with prodromal symptoms occurring 2 years before. His past medical history was negative for peripheral mononeuropathy as well as allergic disease, including allergic rhinitis, sinusitis, and bronchial asthma. He was a non-smoker and did not consume alcohol/supplements/herbals. He also denied previous or ongoing use of drugs. Physical examination was unremarkable except for signs of jaundice and body itching. Complete blood count findings were remarkable for an elevated absolute eosinophil count (AEC) of 2592 cells/μL. Blood chemistry findings included serum total protein, 11.71 g/dL; serum albumin, 2.54 g/dL; total bilirubin, 8.10 mg/dL; direct bilirubin, 6.48 mg/dL, serum aspartate aminotransferase (AST), 385 IU/L; alanine aminotransferase (ALT), 429 IU/L; alkaline phosphatase (ALP), 123 IU/L; and prothrombin time-international normalized ratio, 1.11. Serologic tests were negative for infection with Human Immunodeficiency Virus as well as hepatitis A, B, C or E. The patient’s autoantibody testing (using indirect immunofluorescent method) showed anti-nuclear antibody (ANA) was positive with titer at 1:80 and cytoplasmic pattern while smooth muscle antibody (SMA), anti-mitochondrial antibody and anti-neutrophil cytoplasmic antibodies were negative. The patient’s serum IgG concentration level was 5222 mg/dL (reference range: 548–1768 mg/dL), and his IgG4 concentration level was 1780 mg/dL (reference range: 3.9–86.4 mg/dL). Magnetic resonance imaging (MRI) revealed no abnormalities in the hepatobiliary system or pancreas.
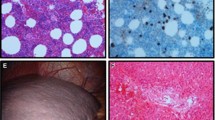
A percutaneous ultrasound-guided liver biopsy was performed (Fig. 1). The pathology results revealed interface hepatitis and bridging necrosis together with severe lymphoplasmacytic cell infiltration, of which most were positive for IgG4 immunostaining (more than 40 IgG4-positive plasma cells/ high power field) in the portal region, with a ratio of IgG4/IgG positive plasma cells greater than 40%. Eosinophilic infiltration was detected with more than 10 cells/high power field. No evidence of bile duct destruction, granuloma, broad collapse or necrotizing vasculitis was noted. Definite AIH was diagnosed based on the patient’s pretreatment revised International Autoimmune Hepatitis Group (IAIHG) score of 17 [5] and the simplified AIH score of 8 [6]. Peripheral blood eosinophilia was diagnosed based on an elevated absolute eosinophil count more than 500cells/μL. Initially, the patient was successfully treated with 40 mg of prednisolone daily for 4 weeks. At his 4-week follow-up visit, the patient exhibited no signs of jaundice, and his liver function tests, and blood eosinophil count eventually normalized. After steroid dose tapering, he continued taking prednisone 5 mg daily to prevent disease recurrence. He decided to receive lifelong prednisolone without undergoing second liver biopsy for disease activity evaluation. His illness did not recur under maintenance therapy of 5 mg prednisolone during the 3-year follow up. His main laboratory results at presentation and during treatment are shown in Table 1.

Photographs of liver biopsy demonstrates (a) The portal tract expanded by severe inflammatory cell infiltration, consisting of lymphocytes and plasma cells (H&E × 100); (b) A moderate number of eosinophils and histiocytes, with interface hepatitis and bridging necrosis, are present; no florid duct lesion or broad collapse is clearly seen (H&E × 400); (c) Immunostaining for IgG4 demonstrates that most plasma cells are marked with IgG4, with more than 40 positive plasma cells/high power field; (d) Immunohistochemical staining with CK7 revealed a moderate number of central and peripheral bile ducts without evidence of destruction (lumen × 200)
Discussion
The IgG4-related disease has recently been defined as a systemic, chronic, relapsing, multiorgan, fibroinflammatory condition. It commonly involves the pancreas, biliary tract, salivary glands, lacrimal glands, and kidneys [2]. The hepatic manifestations of IgG4-related disease are heterogenous and remain unclear [7].
However, it has recently been postulated that there is an association between IgG4-related disease and autoimmune hepatitis, and this has given rise to the disease concepts known as “IgG4 hepatopathy” [4] and “IgG4 associated autoimmune hepatitis” [8]. According to Nakanuma et al., these two types of IgG4-related liver disease can be distinguished clinicopathologically [9]. IgG4 hepatopathy is a collective term for hepatic lesions primarily or secondarily related to sclerosing cholangitis and autoimmune pancreatitis; relevant histological findings include portal inflammation, interface hepatitis, lobular hepatitis, large bile duct obstruction, and canalicular cholestasis [10]. In contrast, IgG4-AIH is clinicopathologically similar to AIH except for marked infiltration of IgG4-positive plasma cells in the liver tissue and elevated serum levels of IgG4 [8]. However, agreed-upon diagnostic criteria have not yet been established. The previously published literature indicates that an IgG4-AIH diagnosis may be based on three features: (1) a finding of “definite AIH” according to the IAIHG scoring system, (2) a serum IgG4 concentration level of at least 135 mg/dL, and (3) an IgG4-expressing plasma cell infiltration of at least 10 cells/high power field in the portal tract [8]. Our patient met all these criteria: his IAIHG score indicated “definite AIH”; his IgG4 level was high, and he had an IgG4/IgG-positive cell ratio greater than 40%. In addition, the findings typical of pancreatic lesion/sclerosing cholangitis were not seen on MRI, nor was there any evidence of portal sclerosis or liver cholestasis. For these reasons, the patient was diagnosed with IgG4-AIH and not IgG4 hepatopathy.
An important question is whether IgG4-AIH is a subtype of classic AIH or a subtype of IgG4-related disease that involved liver. Previous reports suggest that IgG4-AIH should be differentiated from classic AIH [8]. Most baseline biochemical and autoantibody laboratory values do not help distinguish between IgG4-AIH and classic AIH. In contrast, serum concentration levels of both IgG and IgG4 are significantly higher in patients with IgG4-AIH than in those with classic AIH [8, 11]. Histological finding of eosinophilic infiltrate is not a characteristic feature of IgG4-AIH because this could be detected in patients with both group [8]. Both conditions respond dramatically to glucocorticoid therapy. The long-term response to glucocorticoid therapy is comparable for each situation; however, the alanine aminotransferase normalization time after initiation of such treatment is shorter in IgG4-AIH patients [10]. Chung et al. proposed that the degree of accumulation of IgG4-positive liver cells is associated with the serum IgG4 response in patients with IgG4-AIH [11]. Furthermore, synchronous or metachronous development of other IgG4-related disease is observed in most cases of IgG4-AIH [9, 12,13,14,15]. This evidence suggests that IgG4-AIH is a hepatic manifestation of IgG4-related disease and, while sharing pathological findings with AIH, is not a subtype of classic AIH [9]. To our knowledge, this is the second reported case of IgG4-AIH, which had only hepatitis after those reported by Umemura et al. [16].
Another important aspect of our case was the presence of peripheral blood eosinophilia and the abundance of eosinophilic infiltration in the biopsied liver specimen. IgG4 and IgE share a common immune response pathway. Allergic immunology triggers T-helper 2-type immune response that promotes secretion of IgG4 and IgE and induces peripheral blood and tissue eosinophilia [17]. There are data supporting a positive association between IgG4 and both IgE and peripheral eosinophil count [18]. Further, peripheral blood and tissue eosinophilia have been observed in some cases of IgG4-related disease [8]. Moreover, the histologic features of IgG4-related disease often involve increased eosinophil infiltration [2]. Although serum IgE levels were not obtained in this case, our finding of peripheral blood and tissue eosinophilia could be explained by the presence of IgG4-related disease. In a recent study by Mohapatra et al. [3], it was shown that peripheral eosinophilia increased as serum IgG4 increased. This finding was consistent with a recently reported case of IgG4-AIH with the laboratory of IgG4 level is remarkably high (3560 mg/dL) presenting as idiopathic hypereosinophilia syndrome (AEC 17,940 cells/μL) [19]. The author concluded that having peripheral eosinophilia may correspond with having a higher level of serum IgG4. These data are consistent with our findings: our patient had an extremely high level of serum IgG4 (1780 mg/dL) along with significant peripheral eosinophilia (an absolute eosinophil count of 2592 cells/μL). However, the utility of peripheral eosinophilia as a tool for diagnosing IgG4-related disease requires further investigation.
In conclusion, we report a rare case of IgG4-AIH with peripheral blood eosinophilia and absence of symptoms related to other organ of IgG4-related disease. The identification of “Definite AIH” based on both IAIHG and simplified AIH score, high serum IgG4 level, and an accumulation of IgG4-positive cells in the liver were necessary for our diagnosis. Glucocorticoid treatment is the first line treatment for this condition and, in our patient, produced a good response. Although the presence of peripheral eosinophilia may possibly help diagnose this disease, further studies are needed to confirm this and to clarify the clinical course and optimal treatment of IgG4-AIH.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AIH:
-
Autoimmune hepatitis
- IgG4:
-
Immunoglobulin G4
- IgG4-AIH:
-
Immunoglobulin G4associated AIH
- AST:
-
Aspartate aminotransferase
- ALT:
-
Alanine aminotransferase
- IAIHG:
-
International Autoimmune Hepatitis Group
References
Manns MP, Lohse AW, Vergani D. Autoimmune hepatitis--update 2015. J Hepatol. 2015;62(1 Suppl):S100–11.
Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–51.
Mohapatra S, Charilaou P, Sharma A, Singh DP, Sah RP, Murray D, Majumder S, Topazian MD, Chari ST. Significance of peripheral eosinophilia for diagnosis of IgG4-related disease in subjects with elevated serum IgG4 levels. Pancreatology. 2020;20(1):74–8.
Umemura T, Zen Y, Hamano H, Kawa S, Nakanuma Y, Kiyosawa K. Immunoglobin G4-hepatopathy: association of immunoglobin G4-bearing plasma cells in liver with autoimmune pancreatitis. Hepatology. 2007;46(2):463–71.
Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, Chapman RW, Cooksley WG, Czaja AJ, Desmet VJ, et al. International autoimmune hepatitis group report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31(5):929–38.
Clinical Practice Guidelines EASL. Autoimmune hepatitis. J Hepatol. 2015;63(4):971–1004.
Joshi D, Webster GJ. Biliary and hepatic involvement in IgG4-related disease. Aliment Pharmacol Ther. 2014;40(11–12):1251–61.
Umemura T, Zen Y, Hamano H, Joshita S, Ichijo T, Yoshizawa K, Kiyosawa K, Ota M, Kawa S, Nakanuma Y, et al. Clinical significance of immunoglobulin G4-associated autoimmune hepatitis. J Gastroenterol. 2011;46(Suppl 1):48–55.
Nakanuma Y, Ishizu Y, Zen Y, Harada K, Umemura T. Histopathology of IgG4-related autoimmune hepatitis and IgG4-related Hepatopathy in IgG4-related disease. Semin Liver Dis. 2016;36(3):229–41.
Minaga K, Watanabe T, Chung H, Kudo M. Autoimmune hepatitis and IgG4-related disease. World J Gastroenterol. 2019;25(19):2308–14.
Chung H, Watanabe T, Kudo M, Maenishi O, Wakatsuki Y, Chiba T. Identification and characterization of IgG4-associated autoimmune hepatitis. Liver International. 2010;30(2):222–31.
Ishizu Y, Ishigami M, Kuzuya T, Honda T, Hayashi K, Nakano I, Hirooka Y, Goto H. Immunoglobulin G4-associated autoimmune hepatitis later complicated by autoimmune pancreatitis: a case report. Hepatol Res. 2016;46(6):601–6.
Matsumoto K, Kikuchi K, Kuniyoshi N, Tsunashima H, Sekine K, Mabuchi M, Doi S, Zen Y, Miyakawa H. Immunoglobulin G4-related liver disease overlapping with non-alcoholic Steatohepatitis that was diagnosed simultaneously with autoimmune pancreatitis: a case report and review of the literature. Intern Med. 2019;58(24):3537–43.
Li H, Sun L, Brigstock DR, Qi L, Gao R. IgG4-related sclerosing cholangitis overlapping with autoimmune hepatitis: report of a case. Pathol Res Pract. 2017;213(5):565–9.
Nagashima K, Sano I, Kobayashi T, Eto K, Nagai K, Ninomiya R, Suzuki A, Oohata Y, Konishi K, Nakano T, et al. IgG4-related lung Pseudotumor and pleural inflammation with autoimmune hepatitis. Intern Med. 2018;57(1):43–8.
Umemura T, Zen Y, Nakanuma Y, Kiyosawa K. Another cause of autoimmune hepatitis. Hepatology. 2010;52(1):389–90.
Weindorf SC, Frederiksen JK. IgG4-related disease: a reminder for practicing pathologists. Arch Pathol Lab Med. 2017;141(11):1476–83.
Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy. 2014;69(2):269–72.
Kastin D, Siegel M, Anderson R, Aronsohn A. IgG 4 autoimmune hepatitis presenting as idiopathic hypereosinophilia syndrome. Hepatology. 2020. https://doi.org/10.1002/hep.31492.
Acknowledgements
None.
Funding
The authors have no grant to declare for this research from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Conception and design: A.C., V.P.; Review and data collection: A. C, C.C., K. A, A.R.; Draft of the article, provision of table and figures: A. C, C.C.; Study supervision and final approval of the version: V.P.; All authors contributed important intellectual content and approved the final version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was reviewed and approved by the Institutional Review Board of Hatyai Hospital in Songkhla, Thailand (protocol number 54/2563).
Consent for publication
Written informed consent for publication of personal/clinical details and images was obtained from the patient.
Competing interests
The authors declared that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chang, A., Charoenlap, C., Akarapatima, K. et al. Immunoglobulin G4-associated autoimmune hepatitis with peripheral blood eosinophilia: a case report. BMC Gastroenterol 20, 420 (2020). https://doi.org/10.1186/s12876-020-01559-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-020-01559-7