
Abstract
Background
Coronary artery disease (CAD) is a complex disease that is influenced by environmental and genetic factors. In this study, we aimed to investigate the relationship between coding variants in lipid metabolism-related genes and CAD in a Chinese Han population.
Methods
A total of 252 individuals were recruited for this study, including 120 CAD patients and 132 healthy control individuals. Rare and common coding variants in 12 lipid metabolism-related genes (ANGPTL3, ANGPTL4, APOA1, APOA5, APOC1, APOC3, CETP, LDLR, LIPC, LPL, PCSK9 and SCARB1) were detected via next-generation sequencing (NGS)-based targeted sequencing. Associations between common variants and CAD were evaluated by Fisher’s exact test. A gene-based association test of rare variants was performed by the sequence kernel association test-optimal (SKAT-O test).
Results
We found 51 rare variants and 17 common variants in this study. One common missense variant, LIPC rs6083, was significantly associated with CAD after Bonferroni correction (OR = 0.47, 95% CI = 0.29–0.76, p = 1.9 × 10− 3). Thirty-three nonsynonymous rare variants were identified, including two novel variants located in the ANGPTL4 (p.Gly47Glu) and SCARB1 (p.Leu233Phe) genes. We did not find a significant association between rare variants and CAD via gene-based analysis via the SKAT-O test.
Conclusions
Targeted sequencing is a powerful tool for identifying rare and common variants in CAD. The common missense variant LIPC rs6083 confers protection against CAD. The clinical relevance of rare variants in CAD aetiology needs to be investigated in larger sample sizes in the future.
Similar content being viewed by others


Introduction
Coronary artery disease (CAD) is a common chronic inflammatory disease that remains the leading cause of death worldwide [1]. It was estimated that 700,000 people die from CAD in China every year [2]. Lipid disorders are common conditions that involve abnormal levels of lipids in the blood. These disorders are a significant risk factor for the development of CAD. High levels of low-density lipoprotein (LDL) cholesterol, known as “bad” cholesterol, and low levels of high-density lipoprotein (HDL) cholesterol, known as “good” cholesterol, are associated with an increased risk of CAD. Elevated triglyceride levels can promote plaque formation, and high levels of lipoprotein(a) have been shown to increase the risk of CAD. In addition to conventional risk factors such as hypertension, dyslipidaemia, diabetes, obesity and smoking, genetic factors also play an important role in CAD pathogenesis. To reduce the occurrence of CAD, identifying biomarkers responsible for CAD aetiology is important [3].
Genome-wide association studies have identified many variants associated with CAD [4,5,6,7]. GWASs focus on common variants, and these susceptibility variants are always located within intronic or intergenic regions with relatively small effects. Common and rare variants in the coding region that might also be associated with CAD are generally missed.
Due to the increase in throughput and decrease in costs, next-generation sequencing (NGS)-based technology has been shown to be a powerful tool for identifying novel causal mutations associated with Mendelian diseases [8]. Targeted sequencing is a rapid and cost-effective way to detect known and novel variants in selected sets of genes or genomic regions and has been shown to be an efficient technique for screening variants in complex diseases [9, 10]. Targeted sequencing of a subset of genes generates results with quality identical to that of Sanger sequencing [11]. Four rare variants in the coding region of apolipoprotein C3 (APOC3) that disrupt APOC3 function were found to be associated with lower plasma triglyceride levels and a reduced risk of coronary heart disease [12]. Dewey et al. showed that patients carrying inactivating variants in ANGPTL4 had lower triglyceride levels and a lower risk of CAD than noncarriers [13]. Compound heterozygosity for two distinct nonsense variants in ANGPTL3 results in decreased plasma LDL cholesterol levels and familial combined hypolipidaemia [14]. Rare alleles at LDLR and APOA5 confer risk for early-onset myocardial infarction [15]. Rare nonsynonymous variants can facilitate the exploration of disease pathogenesis and provide supportive evidence for putative drug targets for novel therapies.
In this study, we conducted targeted sequencing of 12 genes involved in lipoprotein metabolism to investigate both common and rare variants and their association with CAD. We aimed to identify genetic risk factors that confer susceptibility to CAD in the Chinese Han population, thereby shedding light on the pathogenesis of CAD.
Materials and methods
Study population
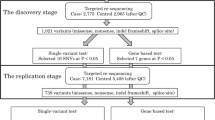
A total of 120 CAD patients and 132 non-CAD control individuals were recruited from Renji Hospital between 2016 and 2020. All of the participants were adults who signed an informed consent form. The average age of the participants was 64.60 ± 9.58 years. All the participants were unrelated Chinese Han individuals. This study was approved by the Medical Ethics Committee of Renji Hospital and complied with the principles set forth by the Declaration of Helsinki. The diagnostic criteria for CAD patients were defined as follows: at least one of the major segments of coronary arteries (right coronary artery, left circumflex, or left anterior descending arteries) with ≥ 50% organic stenosis based on coronary angiography. The clinical characteristics of the CAD patients are summarized in Supplementary Table 1. Non-CAD control individuals were defined as those free from coronary lesions (angiography normal). Individuals with incomplete information were excluded. A 5 ml peripheral blood sample was collected from each subject.
Targeted sequencing
Genomic DNA was extracted using a TianGen DNA Extraction Kit (TianGen Ltd., Beijing, China) following the standard protocol. The DNA concentration and quality were measured using a NanoDrop spectrophotometer (Thermo Scientific, USA). All the purified DNA was stored at -80 °C, and 50ng DNA was used for PCR amplification. We selected lipid metabolism-related genes significantly associated with CAD based on existing genome-wide association studies and case‒control studies. We generated a multiplex PCR panel to capture the coding region of lipid metabolism-related genes. After pre-experimental adjustments, 12 genes were ultimately retained for subsequent experiments (ANGPTL3, ANGPTL4, APOA1, APOA5, APOC1, APOC3, CETP, LDLR, LIPC, LPL, PCSK9 and SCARB1). All the genes were shown to be associated with CAD in previous studies (Table 1). PCR primers (Supplementary Table 2) were designed using Oligo 6.0 and synthesized by Shanghai Free Biotechnology Co., Ltd. (Shanghai, China). Coding regions of the target gene were captured by multiplex PCR followed by adaptor addition. The final panel consisted of 203 amplicons with an average size of 250 bp. Paired-end sequencing (2 × 150) was performed with Illumina NovaSeq sequencing instruments (Novogene, Beijing, China).
Variant analysis
Nonsynonymous exonic variants were identified by BWA (version 0.7.17) and SAMtools (version 1.9) according to the following quality control criteria: (1) at least 50× coverage; (2) Q-score > 30; and (3) at least 40% variant frequency. Variant annotation was performed on GRCh38.p13. Variations that were absent or had a minor allele frequency < 0.01 in the public database (dbSNP build 155; Exome Aggregation Consortium v1.0; 1000 Genomes Project phase 3; and Genome Aggregation Database v3.1.1) were regarded as rare variants. Common variants were defined as having a minor allele frequency > 0.05, and low-frequency variants were defined as having a minor allele frequency between 0.01 and 0.05. Associations between common variants and CAD were determined using a standard Fisher’s exact test with default parameters. The p value was adjusted to 2.9 × 10− 3 by Bonferroni correction (0.05/17). A gene-based association test of rare variants was performed using the sequence kernel association test-optimal (SKAT-O) test [31]. Pathogenicity prediction of the missense variant was performed with SIFT v4.0.3 (http://provean.jcvi.org/index.php) and PolyPhen-2 version 2.2.3 (http://genetics.bwh.harvard.edu/pph2/index.shtml) [32, 33]. Variants were classified into five classes (benign, likely benign, uncertain significance, likely pathogenic, and pathogenic) based on the ACMG guidelines [34]. Multiple sequence alignment was performed using the Clustal method [35].
Results
We screened all the exons of 12 lipid metabolism-related genes (ANGPTL3, ANGPTL4, APOA1, APOA5, APOC1, APOC3, CETP, LDLR, LIPC, LPL, PCSK9 and SCARB1) and their flanking sequences in 120 CAD patients and 132 control individuals. A total of 75 variants were identified after quality control, including 51 rare variants (MAF < 0.01), 7 low-frequency variants (MAF: 0.01–0.05) and 17 common variants (MAF > 0.05).
Seventeen common variants were identified, including 9 synonymous variants and 8 nonsynonymous variants. Common variant association analysis revealed that 4 variants located in the CETP and LIPC genes were nominally associated with CAD (p value < 0.05) (Table 2). Nevertheless, after Bonferroni correction (p value < 2.9 × 10− 3), the missense variant LIPC rs6083 remained significantly associated (p value = 1.9 × 10− 3).
To reveal the potential burden of rare variants in the CAD group compared to the control group, we conducted a gene-based association analysis using the SKAT-O test. However, no significant difference was found between the CAD patients and healthy control individuals (Table 3).
A total of 33 coding nonsynonymous variants, including 32 missense variants and one 7 bp duplication variant, were discovered in 12 gene regions (Table 2). All of these variants were heterozygous variants (Table 4). Of all the rare nonsynonymous variants, 18 were identified only in the CAD group, 12 were identified only in the control group, and 3 were identified in both the CAD and control groups. Of all the rare synonymous variants, 3 were identified only in the CAD group, 10 were identified only in the control group, and 5 were identified in both the CAD and control groups.
Two novel missense variants discovered in this study were not found in the ExAC, gnomAD or dbSNP databases, and both were found in the CAD cohort. One single nucleotide variant in the ANGPTL4 gene that introduces a missense variant at position 47, resulting in the amino variant p.Gly47Glu (GGA-GAA, located in the first exon of ANGPTL4 at nucleotide 8,364,461 on chromosome 19). The other single-nucleotide variant in the SCARB1 gene that introduces a missense variant at position 233, resulting in the amino variant p.Leu233Phe (CTC-TTC, located in the fifth exon of SCARB1 at nucleotide 124,811,899 on chromosome 12). We also identified novel alleles at 4 existing SNVs in the dbSNP database. Two novel variants and four novel alleles were validated by bidirectional Sanger sequencing and demonstrated 100% concordance (Supplementary Fig. 1).
Variant pathogenicity analysis was performed using SIFT and Polyphen-2. Twelve variants were predicted to be deleterious by SIFT, and 18 were predicted to be possibly damaging or probably damaging by PolyPhen-2. Ten variants were predicted to be tolerated by SIFT and benign by PolyPhen-2, indicating that these variants are not pathogenic. Eight variants were predicted to be deleterious/damaging in both programs. Twenty-two variants were predicted to be damaging or deleterious in at least one program. According to the standards and guidelines of the ACMG, p.Asp168Asn in the LDLR gene was classified as a likely pathogenic variant, while the rest were classified as having uncertain significance or benign.
Discussion
In the present study, we systemically screened the coding regions of 12 lipid metabolism-related genes in a Chinese cohort of 120 CAD patients and 132 healthy control individuals. NGS based targeted sequencing can identify not only disease-causing variants but also variants of uncertain significance, which can be challenging for genetic counselling.
We found that the missense variant LIPC rs6083 was associated with protection from CAD. LIPC encodes hepatic triglyceride lipase and participates in the hydrolysis of triglycerides (TGs) and phospholipids [36, 37]. It has been reported that variants in the promoter region of LIPC affect HDL-cholesterol levels [38, 39]. Epigenetic analysis revealed that CAD patients had higher LIPC DNA methylation levels than healthy control individuals [40]. However, further functional verification is needed to determine whether the missense variant LIPC rs6083 affects the pathogenicity of CAD development. Rare variants have a population incidence of < 1% and may not be statistically associated with diseases of interest even in large samples. It was predicted that 27–29% of nonsynonymous variants are neutral or nearly neutral, 30–42% are moderately deleterious, and the remainder are highly deleterious or lethal [41]. Previous studies have shown that rare variants in lipid metabolism genes are associated with CAD. Stitziel et al. reported 37 and 161 loss-of-function variants in 21,980 CAD patients and 158,200 control individuals, respectively. These authors suggested that ANGPTL3 deficiency is associated with protection from CAD (OR = 0.44, p = 0.04) [16]. Cohen et al. reported that 2.6% of black participants (n = 3,363) had nonsense mutations in PCSK9, which was associated with an 88% lower risk of CAD (P = 0.008) [42]. Analysis of the CETP gene revealed that protein truncation variant carrier status was associated with a reduced risk of CAD (OR = 0.70, P = 5.1 × 10 − 3) [25]. To our knowledge, no previous study has reported the relationship between rare variants in lipid metabolism genes and CAD in a Chinese Han cohort. However, there have been association studies of rare variants in other genes with CAD. Jia et al. sequenced nine exons of the CPE gene in 51 CAD patients, and no significant associations were found between rare variants and CAD [43]. Sequencing of MEF2A exon 11 revealed a rare 21-bp deletion in five CAD patients, indicating that this deletion might be a specific cause of CAD [44]. Wang et al. genotyped the rare variant rs34166160 in NINJ2 and demonstrated that rs34166160 significantly confers risk of CAD [45].
In the present study, a total of 33 nonsynonymous rare variants were identified. However, a gene-based SKAT-O test did not reveal an association between rare variants and CAD. We found two novel variants in the CAD cohort. One of them was a variant that introduces a missense variant in ANGPTL4 (p.Gly47Glu), which was predicted to be deleterious by SIFT and probably damaging by PolyPhen-2. The other variant was a variant that introduces a missense variant in SCARB1 (p.Leu233Phe), which was predicted to be tolerated by SIFT and possibly damaged by PolyPhen-2. Protein sequence alignment of the novel variants revealed that both variants affect residues highly conserved across multiple species (Fig. 1). ANGPTL4 inhibits LPL activity and retards lipoprotein catabolism Previous studies have shown that carriers of the ANGPTL4 variant are more likely to have lower triglyceride levels and higher HDL cholesterol levels than noncarriers are and are less likely to have CAD [13]. In this study, the HDL cholesterol levels (1.35 vs. 0.96 ± 0.23) of ANGPTL4 Gly47Glu variant carriers were greater than those of noncarriers, while the TG levels (1.33 vs. 1.59 ± 0.79) were lower. These results are consistent with those of previous studies [13]. Multiple studies have shown that missense variants in the SCARB1 gene are associated with elevated HDL cholesterol levels. Zanoni et al. identified 3 heterozygous carriers and 1 homozygous carrier of p.Pro376Leu through targeted sequencing of SCARB1 in 328 individuals with extremely high plasma HDL cholesterol levels, while the variant did not exist in 398 individuals with extremely low plasma HDL cholesterol levels. Association analysis revealed that carriers of the p.Pro376Leu variant have an increased risk of CAD. (OR = 1.79, P = 0.018) [30]. In this study, individuals carrying the SCARB1 p.Leu233Phe variant had lower-than-average HDL cholesterol levels (0.85 vs. 0.96 ± 0.23).
In the ClinVar database, rs730882109 in LDLR was classified as “conflicting interpretations of pathogenicity”, and rs200727689 in LDLR was classified as “pathogenic/likely pathogenic”. Both of these variants were linked to familial hypercholesterolemia-1 (FHCL1) in the ClinVar database and identified only in the CAD cohort.

Protein sequence alignment across species. A: Amino acid sequence alignment of ANGPTL4 across 11 species. The variant Gly47Glu is indicated by a red box. B: Amino acid sequence alignment of SCARB1 across 11 species. The Leu233Phe variant is indicated by a red box
Conclusion
In summary, we described a novel targeted NGS panel that included 12 lipid metabolism genes. One common missense variant, LIPC rs6083, was significantly associated with a reduced risk of CAD. Of all the rare nonsynonymous variants identified in this study, 18 existed only in the CAD group, 12 were identified only in the control group, and 3 were identified in both groups. However, none of the gene-based SKAT-O tests revealed an association between rare variants and CAD. We identified 33 nonsynonymous rare variants, including two novel variants, one located in the ANGPTL4 gene (p.Gly47Glu) and the other in the SCARB1 gene (p.Leu233Phe). This study suggests that targeted sequencing approaches can be used to discover common and rare variants that contribute to the aetiology of CAD risk and may lead to the discovery of novel pharmaceutical targets for disease prevention and treatment. However, this study has several limitations. (1) This assay was designed to detect single nucleotide variants and small indels, but larger indels or structural rearrangements were missed. (2) Whether these variants alter CAD risk remains unclear due to the lack of statistical power. A larger sample size is needed to increase the statistical power. (3) Determining the pathogenicity of novel variants by computational methods alone is difficult. Functional testing may help to clarify the impact of these variants. (4) As an ageing-related disease, CAD might develop in subjects in the control group in the future, leading to misclassification bias [46]. However, further studies are needed to validate these findings and explore these variations as potential pathogenic variants for CAD.
Data availability
All the data generated or analysed during this study are included in this published article [and its supplementary information files]. All the participants and/or their legal guardian(s) consented to the research process, producing the raw data, tables, and figures for publication. Patient names and patient images are not presented in this study. The datasets generated and/or analysed during the current study are available in the NCBI repository [https://www.ncbi.nlm.nih.gov/sra/PRJNA976403].
Abbreviations
- CAD:
-
Coronary artery disease
- NGS:
-
Next-generation sequencing
References
Roth GA, Abate D, Abate KH, et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the global burden of Disease Study 2017[J]. Lancet. 2018;392(10159):1736–88.
Wang F, Xu C, He Q, et al. Genome-wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population[J]. Nat Genet. 2011;43(4):345–9.
Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation[J]. Nat Rev Genet. 2017;18(6):331–44.
Samani NJ, Erdmann J, Hall AS, et al. Genomewide Association Analysis of coronary artery Disease[J]. N Engl J Med. 2007;357(5):443–53.
Consortium T, W T C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls[J]. Nature. 2007;447(7145):661–78.
Consortium TCG. A genome-wide association study in europeans and South asians identifies five new loci for coronary artery disease[J]. Nat Genet. 2011;43(4):339–44.
Nikpay M, Goel A, Won HH, et al. A comprehensive 1000 genomes–based genome-wide association meta-analysis of coronary artery disease[J]. Nat Genet. 2015;47(10):1121–30.
Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for mendelian disease gene discovery[J]. Nat Rev Genet. 2011;12(11):745–55.
Sadananda SN, Foo JN, Toh MT, et al. Targeted next-generation sequencing to diagnose disorders of HDL cholesterol[J]. J Lipid Res. 2015;56(10):1993–2001.
Safarova MS, Fan X, Austin EE, Arteriosclerosis, et al. Thromb Vascular Biology. 2019;39(6):1227–33.
Sikkema-Raddatz B, Johansson LF, de Boer EN, et al. Targeted next-generation sequencing can replace Sanger sequencing in Clinical Diagnostics[J]. Hum Mutat. 2013;34(7):1035–42.
Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and Coronary Disease[J]. N Engl J Med. 2014;371(1):22–31.
Dewey FE, Gusarova V, O Dushlaine C, et al. Inactivating variants in ANGPTL4 and risk of coronary artery Disease[J]. N Engl J Med. 2016;374(12):1123–33.
Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing,ANGPTL3 mutations, and familial combined Hypolipidemia[J]. N Engl J Med. 2010;363(23):2220–7.
Do R, Stitziel NO, Won H, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction[J]. Nature. 2015;518(7537):102–6.
Stitziel NO, Khera AV, Wang X, et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease[J]. J Am Coll Cardiol. 2017;69(16):2054–63.
Stitziel NO, Stirrups KE, Masca NG, et al. Coding variation inANGPTL4,LPL, and SVEP1 and the risk of Coronary Disease[J]. N Engl J Med. 2016;374(12):1134–44.
Helgadottir A, Gretarsdottir S, Thorleifsson G, et al. Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease[J]. Nat Genet. 2016;48(6):634–9.
Wang F, Wang IZ, Ellis S, et al. Analysis of causal effect of APOA5 variants on premature coronary artery disease[J]. Ann Hum Genet. 2018;82(6):437–47.
Soufi M, Sattler AM, Kurt B, et al. Mutation screening of the APOA5 gene in subjects with coronary artery Disease[J]. J Investig Med. 2015;60(7):1015–9.
Goyal S, Tanigawa Y, Zhang W, et al. APOC3 genetic variation, serum triglycerides, and risk of coronary artery disease in Asian indians, europeans, and other ethnic groups[J]. Lipids Health Dis. 2021;20(1):113.
Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, et al. Loss-of-function mutations inAPOC3 and risk of ischemic vascular Disease[J]. N Engl J Med. 2014;371(1):32–41.
Boekholdt SM, Kuivenhoven J, Hovingh GK, et al. CETP gene variation: relation to lipid parameters and cardiovascular risk[J]. Curr Opin Lipidol. 2004;15(4):393–8.
Zhong S, Sharp DS, Grove JS, et al. Increased coronary heart disease in Japanese-American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels.[J]. J Clin Invest. 1996;97(12):2917–23.
Nomura A, Won H, Khera AV, et al. Protein-truncating variants at the Cholesteryl Ester Transfer Protein Gene and risk for Coronary Heart Disease[J]. Circul Res. 2017;121(1):81–8.
Ference BA, Kastelein JJP, Ray KK, et al. Association of triglyceride-lowering LPL variants and LDL-C–Lowering LDLR variants with risk of Coronary Heart Disease[J]. JAMA. 2019;321(4):364.
Baroni MG, Berni A, Romeo S, Genetic study of common variants at the Apo E, Apo AI, Apo CIII, Apo B et al. lipoprotein lipase (LPL) and hepatic lipase (LIPC) genes and coronary artery disease (CAD): variation in LIPC gene associates with clinical outcomes in patients with established CAD[J]. BMC Medical Genetics, 2003,4(1):8.
Khera AV, Won H, Peloso GM, et al. Association of Rare and Common Variation in the lipoprotein lipase gene with coronary artery Disease[J]. JAMA. 2017;317(9):937.
Koenig SN, Sucharski HC, Jose EM, et al. Inherited variants in SCARB1 cause severe early-onset coronary artery Disease[J]. Circul Res. 2021;129(2):296–307.
Zanoni P, Khetarpal SA, Larach DB, et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease[J]. Science. 2016;351(6278):1166–71.
Wu MC, Lee S, Cai T, et al. Rare-variant association testing for sequencing data with the sequence kernel association test[J]. Am J Hum Genet. 2011;89(1):82–93.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of human missense mutations using PolyPhen-2[J]. Curr Protocols Hum Genet. 2013;76(1):t7–t20.
Choi Y, Sims GE, Murphy S, et al. Predicting the functional effect of amino acid substitutions and Indels[J]. PLoS ONE. 2012;7(10):e46688.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Sci. 2015;17(5):405–24.
Higgins DG, Sharp PM. CLUSTAL: a package for performing multiple sequence alignment on a microcomputer[J]. Gene. 1988;73(1):237–44.
Kobayashi J, Miyashita K, Nakajima K, et al. Hepatic lipase: a comprehensive view of its role on plasma lipid and lipoprotein Metabolism[J]. J Atheroscler Thromb. 2015;22(10):1001–11.
Annema W, Tietge UJ. Role of hepatic lipase and endothelial lipase in high-density lipoprotein-mediated reverse cholesterol transport[J]. Curr Atheroscler Rep. 2011;13(3):257–65.
Isaacs A, Sayed-Tabatabaei FA, Njajou OT, et al. The – 514 C->T hepatic lipase promoter region polymorphism and plasma lipids: a meta-analysis[J]. J Clin Endocrinol Metab. 2004;89(8):3858–63.
Hodoglugil U, Williamson DW, Mahley RW. Polymorphisms in the hepatic lipase gene affect plasma HDL-cholesterol levels in a Turkish population[J]. J Lipid Res. 2010;51(2):422–30.
Li W, Wang Y, Huang R, et al. Association of lipid metabolism-related gene promoter methylation with risk of coronary artery disease[J]. Mol Biol Rep. 2022;49(10):9373–8.
Boyko AR, Williamson SH, Indap AR, et al. Assessing the evolutionary impact of amino acid mutations in the Human Genome[J]. PLoS Genet. 2008;4(5):e1000083.
Cohen JC, Boerwinkle E, Mosley TH, et al. Sequence variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease[J]. N Engl J Med. 2006;354(12):1264–72.
Jia E, Wang J, Yang Z, et al. Molecular scanning of the human carboxypeptidase E gene for mutations in Chinese subjects with coronary atherosclerosis[J]. Mol Cell Biochem. 2007;307(1–2):31–9.
Liu Y, Niu W, Wu Z, et al. Variants in exon 11 of MEF2A gene and coronary artery disease: evidence from a case-control study, systematic review, and Meta-Analysis[J]. PLoS ONE. 2012;7(2):e31406.
Wang P, Wang Y, Peng H, et al. Functional rare variant in a C/EBP beta binding site in NINJ2 gene increases the risk of coronary artery disease[J]. Aging. 2021;13(23):25393–407.
Mitchell BD, Fornage M, McArdle PF, et al. Using previously genotyped controls in genome-wide association studies (GWAS): application to the Stroke Genetics Network (SiGN)[J]. Front Genet. 2014;5:95.
Acknowledgements
We thank Hu Liu from Shanghai Lehao Bio-Science Company for his technical support with the gene sequencing.
Funding
This research was funded by the Shanghai Science and Technology Development Foundation (SY20221RUE01).
Author information
Authors and Affiliations
Contributions
Wei Li, Yongyi Wang and Ritai Huang performed the annotation analysis, produced figures, and participated in drafting the manuscript. Feng Lian,Weijun Wang and Genxing Xu provided the data interpretation and offered the study advice. Song Xue designed and supervised the project, generated results, provided data interpretation. All authors read, revised, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Shanghai Jiaotong University School of Medicine, Renji Hospital Ethics Committee (KY2021-159-B), and complied with the principles set forth by the Declaration of Helsinki. All participants or their guardians were aware of the purpose, risks and rights of this research. Their personal safety and health rights have been fully guaranteed. All participants were free to withdraw at any time, and their treatment activities were not affected by the withdrawal. Informed consent was signed by all participants. The researchers followed the requirements of the GCP and ethical laws and regulations and strictly followed the approved protocols and documents to carry out this clinical study. All research involving human genetic resource activities was approved/recorded by the Office of Human Genetics and carried out after the start of ethical approval.
Consent for publication
The manuscript does not involve any identifiable images or data. Consent for publication is not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, W., Wang, Y., Huang, R. et al. Rare and common coding variants in lipid metabolism-related genes and their association with coronary artery disease. BMC Cardiovasc Disord 24, 97 (2024). https://doi.org/10.1186/s12872-024-03759-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-024-03759-5