
Abstract
Background
The MYH7 gene, which encodes the slow/ß-cardiac myosin heavy chain, is mutated in myosin storage myopathy (MSM). The clinical spectrum of MSM is quite heterogeneous in that it ranges from cardiomyopathies to skeletal myopathies or a combination of both, depending on the affected region. In this study, we performed clinical and molecular examinations of the proband of an Iranian family with MSM in an autosomal dominant condition exhibiting proximal muscle weakness and dilated cardiomyopathy.
Methods
Following thorough clinical and paraclinical examinations, whole-exome sequencing `was performed on the proband (II-5). Pathogenicity prediction of the candidate variant was performed through in-silico analysis. Co-segregation analysis of the WES data among the family members was carried out by PCR-based Sanger sequencing.
Results
A novel heterozygous missense variant, MYH7 (NM_000257): c.C1888A: p.Pro630Thr, was found in the DNA of the proband and his children and confirmed by Sanger sequencing. The in-silico analysis revealed that p.Pro630Thr substitution was deleterious. The novel sequence variant fell within a highly conserved region of the head domain. Our findings expand the spectrum of MYH7 mutations.
Conclusions
This finding could improve genetic counseling and prenatal diagnosis in families with clinical manifestations associated with MYH7-related myopathy.
Similar content being viewed by others

Introduction
Myosin, which is a highly conserved protein in all eukaryotic cells, is not only the major constituent of skeletal muscle thick filaments but also a crucial element for body movement and heart contractility. It contains two elongated globular heads connected to a long helical coiled coil (the myosin rod). This hexameric protein consists of two myosin heavy chain (MyHC) subunits and four light chain subunits. Each head, or subfragment 1 (S1), is comprised of approximately 850 N-terminal residues of one MyHC and one of each light chain. The heads, including actin and ATP-binding regions, are liable for the force transduction properties of myosin [1]. The N-terminal region of the myosin rod, designated as subfragment 2 (S2), joins S1 to the filament backbone. The myosin rod is a parallel α-helical coiled-coil dimer of the C-terminal of MyHC tails. The larger C-terminal part of the rod, named “light meromyosin (LMM)”, lies along the thick filament axis and mediates filament assembly [2]. The LMM also provides sites for the binding of myosin-associated proteins like myomesin 1, myosin-binding protein C and H, M-protein, and titin.
There are three major MyHC isoforms expressed in human limb skeletal muscles. MyHC IIx, coded by MYH1 as a member of the MYH gene family, is expressed in fast, glycolytic, type 2B muscle fibers. MyHC IIa, encoded by MYH2, is expressed in fast, intermediate, type 2 A muscle fibers. Slow/ß-cardiac MyHC (MyHC I), encoded by the MYH7 gene, is expressed in slow, oxidative, type 1 muscle fibers. It is also expressed in the ventricles of the heart [3]. Located on chromosome 14, the MYH7 gene (OMIM # 160,760) contains approximately 22,883 bp, including 41 exons [4]. Pathogenic mutations in MYH7 have been reported to cause a wide range of clinical expressions ranging from hereditary skeletal muscle diseases, including Laing distal myopathy [5] and myosin storage myopathy (MSM) with or without cardiac involvement, to isolated cardiomyopathies such as dilated cardiomyopathy [6], hypertrophic cardiomyopathy [7], and left ventricular non-compaction cardiomyopathy [8], depending on the residue of MYH7 that is affected [9]. MYH7 mutations are reported in 14–25% of all cardiomyopathy cases [10].
MSM (OMIM #608,358), also known as “hyaline body myopathy”, is a rare, congenital myopathy identified by subsarcolemmal accumulations of myosin in type 1 skeletal muscle fibers resulting in the weakness of the scapula, limb, and distal muscles. This myopathy was first described by Cancilla et al. [11] as “familial myopathy with probable lysis of myofibrils of type I fibers” in 1971. Following the molecular nosologic identification of the mutation Arg1845Trp in the rod region of MYH7, Tajsharghi et al. [1] proposed the unifying term “myosin storage myopathy” for this disease in 2003. Although the disease is inherited in an autosomal dominant or recessive fashion [12,13,14], a few sporadic cases with no previous family history have been reported [1, 15]. The onset is generally in childhood, but it may be manifested much later in middle age. Mutations causing MSM are located in the distal end of the tail of MyHC I (exons 37–40 of MYH7) [3]. The clinical manifestations of the disease are highly variable among affected individuals, ranging from no weakness to severe impairment of ambulation [1, 9, 11, 15,16,17,18,19,20,21,22,23] even within the same family [9, 17, 20]. Further, it has been reported that many patients with MSM suffer from delayed motor milestones and usually present with proximal muscle weakness in the four limbs, difficulties in climbing stairs, or running and waddling gait [24].
Given that the conventional approaches to the study of gene mutations are time-consuming and costly, currently, the next-generation sequencing [25]-based method has been widely used to identify the causative variants of many single-gene disorders. We herein describe an Iranian family with an autosomal dominantly inherited pattern of MSM presenting with slowly progressive muscle weakness and dilated cardiomyopathy associated with the MYH7 (NM_000257): c.C1888A: p.Pro630Thr disclosed by whole-exome sequencing. This family remarkably widens the genotypic and phenotypic variability of MSM, manifesting the first report of this variant in MYH7-related myopathy with a somewhat distinct phenotype from Iran. The identification of disease-causing variants in a particular population plays an important role in the development of the molecular diagnosis of such disorders.
Materials and methods
Ethics approval and consent to participate
The present study was performed in accordance with the Declaration of Helsinki and approved by Rajaie Cardiovascular Medical and Research Center (approval number: IR.RHC.REC.1399.019). Written informed consent was obtained from all participants for their participation and publication of this report.
Family recruitment and clinical presentations
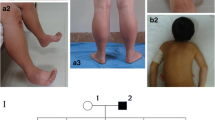
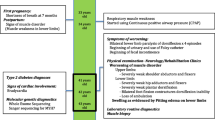
Three generations of an Iranian family recruited for this study are presented in Fig. 1A. The proband (II-5) was a 51-year-old man, who was described as being healthy until age 47. He stated that he had experienced signs of slowly progressive muscle weakness, heart rhythm problems, and extreme shortness of breath since age 47 years. These were worsened during the following four years. Despite the presence of muscle weakness, fatigue, and exercise intolerance with respiratory distress, he had no accurate neuromuscular or cardiological investigation up to the age of 50. A history of taking a statin, which was discontinued, was reported by him. The proband (II-5) had an asymptomatic daughter (Fig. 1A: III-1, 21 years of age), an asymptomatic son (Fig. 1A: III-2, 15 years of age), and also four siblings (II-1, II-2, II-3, and II-4 aged 64, 61, 58, and 55 years, respectively) all without symptoms of muscle or cardiac disease. Unfortunately, the individuals II-1, II-2, II-3, and II-4 were not available to study the clinical and genetic status. Physical examination showed the presence of proximal weakness in the lower limbs and mild scapular winging. He also had some difficulty climbing stairs and lifting his arms above his head. In terms of diagnostic studies, at 51 years of age, the creatine phosphokinase level was elevated to 634 U/L (reference: 0–195). Additionally, high levels of lactate dehydrogenase (295 U/L; normal: 135–225 U/L) and aldolase (7.5 IU/L; normal: 1.5–8.1 IU/L) were found in the proband (II-5). The proband (II-5) was positive for Mi-2a (1+), PL-7 (2+), and RO-52 (2+) in a specific myositis panel. He underwent an extensive clinical investigation, including Pompe disease (MIM #232,300) screening, spiral multi-slice lung computed tomography (CT) scanning, electromyography/nerve conduction studies, muscle biopsies, and magnetic resonance imaging (MRI) on both thighs. Muscle biopsy was performed by open technique and the sample was frozen in isopentane cooled in liquid nitrogen. Frozen sections were stained by Hematoxylin and eosin, Modified Gomori Trichrome, PAS, PAS + diastasis, Oil red O, Congo red, NADH-TR, SDH, COX, COX + SDH and ATPase reactions. These workups led to a diagnosis of myopathy in the proband (II-5).

The image presents pedigree, sequencing chromatograms, myosin structure, and conservation analysis in a family affected by MYH7 mutation. (A) Family pedigree of an Iranian family with myopathy: The family investigated in this study consists of 3 generations and 17 members. Only the proband (II-5) is affected (pointed with an arrow). (B) The snapshot of the sequencing reads: The proband (II-5), his daughter (III-1), and his son (III-2) carry the c.C1888A: p.Pro630Thr variant in a heterozygous status. The black arrow shows the location of the mutated nucleotide. (C) The illustration of the myosin domains. (D) The 3D structure of the native and mutated myosin was built using UniProt (https://www.uniprot.org/). The location of the p.Pro630Thr variant is shown on the head portion of myosin. (E) The evaluation of the amino-acid evolutionary conservation using CLUSTALW (https://www.genome.jp/tools-bin/clustalw): As depicted, the position of this mutation is highly conserved during evolution
Whole-exome sequencing
To determine an accurate mutational diagnosis of myopathy in this family, WES was implemented just on the proband (II-5). Exome was captured using the Agilent SureSelect Exome Capture kit (Agilent Inc, Santa Clara, California, USA). Then, the sequencing of the enriched exon libraries was performed on the Illumina HiSeq 4000 (Macrogen Inc, Seoul, South Korea). The sequencing reads were aligned to the human genome reference (GRCh37 build) by the BWA (v07.17) tool [26]. Next, single-nucleotide polymorphisms/small insertion and deletion (SNP/InDel) was called by applying the GATK (v4.1.4.1) tool with the result file of mapping (BAM). Marking and removing duplicates were performed by SAMtools (in GATK package) [27], followed by recalibration and SNP/InDel calling. For filtering and prioritization, the variants with a minor allele frequency (MAF) more than 0.05 in the 1000 Genomes Project, gnomAD (v2.1.1), and ExAC databases [25] were removed. Prediction tools such as CADD, SIFT, PolyPhen-2, PROVEAN, FATHMM, and GERP++ were used for predictive analytics. Accordingly, the variant that was interpreted to be pathogenic in at least four algorithms was considered for confirmation/segregation analysis.
Polymerase chain reaction (PCR) and segregation analysis
The variant of the MYH7 gene was sequenced by the PCR and Sanger sequencing method. The primer pairs were designed and validated using Primer3 (v.04.0) (http://bioinfo.ut.ee/primer3-0.4.0/) and BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome). forward: 5’TATATTGACCATAGAGCAGAA3’ and reverse: 5’TTGCCCTTCTCAATAGCTGCAG3’. PCR was performed on a SimpliAmp™ Thermal Cycler (Thermo Fisher Scientific), with 100 ng DNA, 10pmol/L of primers, 1.5 mmol/L of MgCl2, 200 mmol/L of dNTP, and 1 U Taq DNA polymerase (Amplicon, UK). Then incubation was carried out at 95 °C for 5 min, 35 amplification cycle (30 S at 95 °C, 30 S at 62 °C, and 30 S at 72 °C). Sanger sequencing was done using the BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies; Thermo Fisher Scientific, Shanghai, China) on the ABI Sequencer 3500XL PE (Applied Biosystems, CA, USA).
Results
Pompe disease evaluation/screening
At age 51, the α-1, 4-glucosidase activity of the proband (II-5) was 6.7 in units of µmol/L/h, above the cut-off value (> 2.0). Therefore, Pompe disease was unlikely in the proband (II-5).
MRI and CT scan findings
The MRI findings of the proband (II-5), performed at age 51, showed dilated cardiomyopathy. The proband (II-5) also underwent a CT scan of his lungs at age 51. Minimal ground-glass opacities were evident in the lower lobes bilaterally, with prominence on the right side. There was no evidence of other pathologic findings in the parenchyma of both lungs. Mild pleural effusion was seen bilaterally, with prominence on the right side. MRI on both thighs, performed at age 51, showed diffuse atrophy and fat deposition in the semitendinosus, semimembranosus, biceps femoris, and soleus muscles.
Electrodiagnostic findings
The results of the electrodiagnostic testing of the proband (II-5), performed at age 51, are presented in Table 1 A-D. Based on the provided nerve conduction study (NCS) and electromyography (EMG) results indicate diminished amplitudes, mildly decelerated conduction velocities, and increased insertional activity, positive sharp waves, and fibrillation potentials in numerous muscles. So, the test concluded a chronic myopathic process with ongoing active denervation with some myotonic discharges.
Pathology
Cytological evaluation of the pleural effusion of the proband (II-5) revealed some reactive and bland-looking isolated and clustered mesothelial cells admixed with some lymphocytes and red blood cells in a proteinaceous background. No malignant cells were detected. A muscle biopsy taken from the left vastus lateralis at age 51 years demonstrated mild myopathic atrophy. Hematoxylin and eosin staining showed striated muscle tissue with variation in fiber size. Atrophic fibers were round or angular and dispersed (Fig. 2A). Rare dispersed necrotic fibers were seen (Fig. 2B). Only one fiber showed a subsarcolemmal aggregate of homogenous basophilic materials. Internalized nuclei were increased (Fig. 2C). There was neither fibrosis nor inflammation. Modified Gomori trichrome stain revealed a few ragged red fibers and rare red-rimmed cytoplasmic vacuoles (Fig. 2D). NADH-TR reaction demonstrated good differentiation of muscle fibers with slight nonspecific intermyofibrillar network abnormalities as some uneven cytoplasmic staining.SDH reaction illustrated a few fibers with abnormal mitochondrial proliferation. COX + SDH reaction revealed no COX-negative fiber (Fig. 2E). ATPase reactions PH 9.4, 4.63, and 4.35 revealed type 2 fiber predominance, and no fiber type grouping was seen. Atrophic fibers were mostly type 1, but no fiber type disproportion was detected (Fig. 2F).

The image shows a muscle biopsy from the left vastus lateralis. (A) Fiber size variation with round and angular atrophic fibers and increased internalization of nuclei (H&E X200). (B) One necrotic fiber with myophagocytosis (H&E X200). (C) Subsarcolemmal basophilic aggregate in only one fiber (H&E X200). (D) Red-rimmed vacuole (Modified Gomori Trichrome X200). (E) Checkerboard pattern with no COX-negative fiber (COX + SDH x100). (F) Type 2 fibers predominance and slight atrophy with no fiber type grouping (ATPase 9.4 × 100)
Genetic investigations
After the filtration of the WES data, a novel heterozygous missense mutation in exon 16 of the MYH7 (NM_000257): c.C1888A: p.Pro630Thr, was identified, which was probably responsible for MSM with dilated cardiomyopathy in this family. Sanger sequencing confirmed the presence of the c.C1888A variant in the affected proband (II-5). Both his unaffected daughter (Fig. 1A: III-1) and unaffected son (Fig. 1A: III-2) were heterozygous for this locus (Fig. 1B). Position 630 in the MYH7 protein is highly conserved among multiple species (Fig. 1E), and the missense mutation results in an amino-acid substitution from Proline to Threonine at this position.
A schematic illustration of the myosin protein and its domains is presented in Fig. 1C. The p.Pro630Thr mutation occurred within the head domain. The 3D structures of the protein representing the wild type in contrast with the mutated p.Pro630Thr are depicted in Fig. 1D. According to the American College of Medical Genetics and Genomics 2015 (ACMG) [28], c.C1888A is determined as a likely pathogenic variant (i.e., Criteria: PVS1, PM1, PM2, and PP2). The missense mutation was supported as the cause of the disease by CADD, SIFT, PolyPhen-2, PROVEAN, FATHMM, and GERP++.
Discussion
Mutations in MYH7 encoding for the β-MyHC are a common cause of hypertrophic or dilated cardiomyopathy, Laing distal myopathy, and MSM. MYH7 maps in tandem on human chromosome 14 with MYH6. The MYH7 gene is composed of 40 exons. In particular, mutations that cause MSM are located in exons 37–40 of MYH7 [29, 30].
In this study, we analyzed three generations of an Iranian family with suspected myopathy using WES to detect the causative mutation. We found that the proband (II-5) in the family carried a heterozygous c.C1888A: p.Pro630Thr variant in the MYH7 gene associated with MSM. The proband (II-5) had two children. The two unaffected siblings, III-1 (the proband’s daughter) and III-2 (the proband’s son), carried the same c.C1888A: p.Pro630Thr variant in the MYH7 gene. However, no symptoms of the disease have been witnessed in them thus far, highlighting the importance of discussing disease penetrance during genetic counseling. There was no family history of muscle weakness in the remaining family members. The findings suggested that the c.C1888A: p.Pro630Thr variant could be a de novo variant that appeared to have occurred in the affected proband (II-5). Nonetheless, because DNA was not available from his deceased parents, we could not prove it. The recurrent independent emergence of MYH7 mutations in different ethnic backgrounds is thought to be associated with the high prevalence of de novo mutations in the MYH7 gene [31, 32].
Age at the onset of initial symptoms is usually infancy or childhood with variable penetrance. Still, it has been reported that in some patients, symptoms emerge in adult life or some may even be asymptomatic in their 40s. Li et al. reported two cases including a 46-year-old man with late-onset proximal weakness and his 26-year-old son showing talipes cavus and calf pseudohypertrophy [33]. Bohlega et al. reported three generations of a Saudi Arabian family, with the index patient who experienced the first symptoms around age 40 and her offspring in early childhood [17]. These reports indicate the intrafamilial heterogeneity on both clinical manifestations and age at onset. In the present study, the proband (II-5), a 51-year-old man, manifested his initial symptoms at around 47 years of age with no other affected relatives. Although cardiomyopathy is typically not present in MSM, we observed dilated cardiomyopathy in the proband (II-5).
Predominantly, mutations existing within the globular head of MyHC I have been associated with hypertrophic and dilated cardiomyopathies [34], whereas mutations located in the distal rod region of the protein, including Leu1793Pro, Arg1845Trp, Glu1886Lys, and His1901Leu, have been reported to cause MSM [3, 35]. Nevertheless, this final distinction is not very pertinent since there have been several reports of cases with skeletal myopathies and mutations in the globular head region [36], often representing associated cardiac involvement [9, 37]. On the other hand, numerous reports have described cardiomyopathy and mutations in the COOH-tail region of the protein [3, 14, 38, 39]. Of note, the c.C1888A: p.Pro630Thr variant, which we detected in our study and deemed culpable for MSM, is located in the myosin globular head domain.
Remarkably, different phenotypes have been found to be associated with various mutations of the same amino-acid residue of β-MyHC [32]. The missense mutation, p.Leu1793Pro, is known to cause MSM [12], whereas the heterozygous deletion at this position (pLeu1793del) was reported in a boy with distal myopathy who had undergone heart transplantation at age 3 [40]. Contrarily, the same mutation at the residue can lead to either MSM or Laing early-onset distal myopathy [23, 41]. While it remains largely unexplained why myopathy associated with MYH7 mutations presents a variable phenotypic expression, Tasca et al. suggested that it could be due to the effect of genetic or environmental modifiers [41]. For instance, skeletal muscle fiber type proportions in humans are different based on race [42] and are influenced by both environmental and inherited factors [43]. Differences in disease severity and phenotypes can also result in variation in the ratio of mutant-to-wild type protein [44]. Proteins that interact with myosin tail like titin, alpha actinin, myomesin, M-protein, and desmin show candidate genes to modulate MSM clinical phenotypes and genetic penetrance [9].
A drosophila MSM model has recently been described to study the effects of Leu1793Pro, Arg1845Trp, and Glu1883Lys MSM mutant myosins expressed in an indirect flight and jump muscle myosin null background. Mutant animals showed highly compromised jump and flight ability. The indirect flight structure displayed myofibrillar disarray and degeneration with hyaline-like inclusions. It was demonstrated that the mutant myosin had both decreased ability to polymerize and reduced stability [45, 46]. Dahl-Halvarsson et al. expressed mutated myosin proteins in cultured human muscle cells to evaluate the impact of four missense mutations—namely Leu1793Pro, Arg1845Trp, Glu1883Lys, and His1901Leu—on myosin assembly and muscle function and assess the mechanisms leading to protein aggregation in MSM. The results indicated that the Arg1845Trp and His1901Leu mutants were prone to the formation of myosin aggregates without assembly into striated sarcomeric thick filaments [47]. On the whole, available data suggest that changes in the structural, rather than functional, properties of MyHC I caused by a mutation in the MYH7 gene may exhibit the primary trigger of MSM [47]. Further research is needed to explain the pathogenic basis of MSM, which may play a crucial role in clinical decision-making as well as diagnostic and therapeutic development.
Clinical and genetic characteristics of at-risk individuals and carrier screening can provide more information about genetic counseling for future pregnancies in this family which will improve their quality of life. Moreover, a population study for the frequency of the p.Pro630Thr variant is currently still required. The limitations of our study are the lack of clinical and genetic data of individuals at risk of having inherited the MYH7 variant within the family and also the confirmed carriers.
Conclusions
An accurate diagnosis of myopathy requires information from muscle MRI and/or muscle biopsies, in tandem with complete examinations of clinical phenotypes as well as respiratory and cardiac evaluations. In the last decade, the NGS molecular technology has provided a greater discovery power to detect novel or rare variants even if clinical information is limited. In the present study, WES enabled us to make a possibly diagnosis of myopathy as MSM caused by a mutation in the MYH7 gene.
Data Availability
The datasets generated and/or analyzed during the current study are available in the ClinVar repository[https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV001023003.3/?redir=vcv]. The submission ID of the variant in ClinVar is as follows: NM_000257.4 (MYH7): c.1888 C > A (p.Pro630Thr): VCV001023003.3.
References
Tajsharghi H, Thornell LE, Lindberg C, Lindvall B, Henriksson KG, Oldfors A. Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 2003;54(4):494–500.
Blair E, Redwood C, de Jesus Oliveira M, Moolman-Smook J, Brink P, Corfield V, Ostman-Smith I, Watkins H. Mutations of the light meromyosin domain of the β-myosin heavy chain rod in hypertrophic cardiomyopathy. Circul Res. 2002;90(3):263–9.
Tajsharghi H, Oldfors A. Myosinopathies: pathology and mechanisms. Acta Neuropathol. 2013;125(1):3–18.
Ghazanfari-Sarabi S, Rayati M, Hashemi-Soteh MB. Laing early-onset distal myopathy due to the MYH7 mutation in an iranian family. J Pediatr Rev. 2020;8(1):35–46.
Carbonell-Corvillo P, Tristan-Clavijo E, Cabrera-Serrano M, Servián-Morilla E, García-Martín G, Villarreal-Pérez L, Rivas-Infante E, Area-Gómez E. Chamorro-Muñoz M, Gil-Gálvez A: a novel MYH7 founder mutation causing Laing distal myopathy in Southern Spain. Neuromuscul Disord. 2018;28(10):828–36.
Abdallah AM, Carlus SJ, Al-Mazroea AH, Alluqmani M, Almohammadi Y, Bhuiyan ZA, Al-Harbi KM. Digenic inheritance of LAMA4 and MYH7 mutations in patient with infantile dilated cardiomyopathy. Medicina. 2019;55(1):17.
Goel N, Huddleston CB, Fiore AC. A novel mutation of the MYH7 gene in a patient with hypertrophic cardiomyopathy. Turk J Pediatr. 2018;60(3):315–8.
Kolokotronis K, Kühnisch J, Klopocki E, Dartsch J, Rost S, Huculak C, Mearini G, Störk S, Carrier L, Klaassen S. Biallelic mutation in MYH7 and MYBPC3 leads to severe cardiomyopathy with left ventricular noncompaction phenotype. Hum Mutat. 2019;40(8):1101–14.
Uro-Coste E, Arné-Bes M-C, Pellissier J-F, Richard P, Levade T, Heitz F, Figarella-Branger D, Delisle M-B. Striking phenotypic variability in two familial cases of myosin storage myopathy with a MYH7 Leu1793pro mutation. Neuromuscul Disord. 2009;19(2):163–6.
Sedaghat-Hamedani F, Kayvanpour E, Tugrul OF, Lai A, Amr A, Haas J, Proctor T, Ehlermann P, Jensen K, Katus HA. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: a meta-analysis on 7675 individuals. Clin Res Cardiol. 2018;107(1):30–41.
Cancilla P, Kalyanaraman K, Verity M, Munsat T, Pearson C. Familial myopathy with probable lysis of myofibrils in type I fibers. Neurology. 1971;21(6):579–9.
Dye DE, Azzarelli B, Goebel HH, Laing NG. Novel slow-skeletal myosin (MYH7) mutation in the original myosin storage myopathy kindred. Neuromuscul Disord. 2006;16(6):357–60.
Tajsharghi H, Oldfors A, Macleod DP, Swash M. Homozygous mutation in MYH7 in myosin storage myopathy and cardiomyopathy. Neurology. 2007;68(12):962–2.
Yüceyar N, Ayhan Ö, Karasoy H, Tolun A. Homozygous MYH7 R1820W mutation results in recessive myosin storage myopathy: scapuloperoneal and respiratory weakness with dilated cardiomyopathy. Neuromuscul Disord. 2015;25(4):340–4.
Ceuterick C, Martin J, Martens C. Hyaline bodies in skeletal muscle of a patient with a mild chronic nonprogressive congenital myopathy. Clin Neuropathol. 1993;12(2):79–83.
Barohn RJ, Brumback RA, Mendell JR. Hyaline body myopathy. Neuromuscul Disord. 1994;4(3):257–62.
Bohlega S, Lach B, Meyer B, Al Said Y, Kambouris M, Al Homsi M, Cupler E. Autosomal dominant hyaline body myopathy: clinical variability and pathologic findings. Neurology. 2003;61(11):1519–23.
Laing N, Ceuterick-de Groote C, Dye D, Liyanage K, Duff R, Dubois B, Robberecht W, Sciot R, Martin J, Goebel H. Myosin storage myopathy: slow skeletal myosin (MYH7) mutation in two isolated cases. Neurology. 2005;64(3):527–9.
Masuzugawa S, Kuzuhara S, Narita Y, Naito Y, Taniguchi A, Ibi T. Autosomal dominant hyaline body myopathy presenting as scapuloperoneal syndrome: clinical features and muscle pathology. Neurology. 1997;48(1):253–7.
Pegoraro E, Gavassini BF, Borsato C, Melacini P, Vianello A, Stramare R, Cenacchi G, Angelini C. MYH7 gene mutation in myosin storage myopathy and scapulo-peroneal myopathy. Neuromuscul Disord. 2007;17(4):321–9.
Sahgal V, Sahgal S. A new congenital myopathy. Acta Neuropathol. 1977;37(3):225–30.
Shingde MV, Spring PJ, Maxwell A, Wills EJ, Harper CG, Dye DE, Laing NG, North KN. Myosin storage (hyaline body) myopathy: a case report. Neuromuscul Disord. 2006;16(12):882–6.
Stalpers X, Verrips A, Braakhekke J, Lammens M, van den Wijngaard A, Mostert A. Scoliosis surgery in a patient with de novo myosin storage myopathy. Neuromuscul Disord. 2011;21(11):812–5.
Dahl-Halvarsson M, Olive M, Pokrzywa M, Norum M, Ejeskär K, Tajsharghi H. Impaired muscle morphology in a Drosophila model of myosin storage myopathy was supressed by overexpression of an E3 ubiquitin ligase. Dis Models Mech 2020, 13(12).
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26(5):589–95.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, Del Angel G, Rivas MA, Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Sci. 2015;17(5):405–23.
Gacita AM, Fullenkamp DE, Ohiri J, Pottinger T, Puckelwartz MJ, Nobrega MA, McNally EM. Genetic Variation in Enhancers Modifies Cardiomyopathy Gene Expression and Progression. Circulation 2021.
Kiphuth I, Neuen-Jacob E, Struffert T, Wehner M, Wallefeld W, Laing N, Schröder R. Myosin storage myopathy: a rare subtype of protein aggregate myopathies. Fortschr Neurol Psychiatr. 2010;78(4):219–22.
Dubourg O, Maisonobe T, Behin A, Suominen T, Raheem O, Penttilä S, Parton M, Eymard B, Dahl A, Udd B. A novel MYH7 mutation occurring independently in french and norwegian laing distal myopathy families and de novo in one finnish patient. J Neurol. 2011;258(6):1157–63.
Clarke NF, Amburgey K, Teener J, Camelo-Piragua S, Kesari A, Punetha J, Waddell LB, Davis M, Laing NG, Monnier N. A novel mutation expands the genetic and clinical spectrum of MYH7-related myopathies. Neuromuscul Disord. 2013;23(5):432–6.
Li N, Zhao Z, Shen H, Bing Q, Guo X, Hu J. MYH7 mutation associated with two phenotypes of myopathy. Neurol Sci. 2018;39(2):333–9.
Walsh R, Rutland C, Thomas R, Loughna S. Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology. 2010;115(1):49–60.
Armel TZ, Leinwand LA. Mutations in the β-myosin rod cause myosin storage myopathy via multiple mechanisms. Proc Natl Acad Sci. 2009;106(15):6291–6.
Homayoun H, Khavandgar S, Hoover JM, Mohsen A-W, Vockley J, Lacomis D, Clemens PR. Novel mutation in MYH7 gene associated with distal myopathy and cardiomyopathy. Neuromuscul Disord. 2011;21(3):219–22.
Darin N, Tajsharghi H, Östman-Smith I, Gilljam T, Oldfors A. New skeletal myopathy and cardiomyopathy associated with a missense mutation in MYH7. Neurology. 2007;68(23):2041–2.
Ruggiero L, Fiorillo C, Gibertini S, De Stefano F, Manganelli F, Iodice R, Vitale F, Zanotti S, Galderisi M, Mora M. A rare mutation in MYH7 gene occurs with overlapping phenotype. Biochem Biophys Res Commun. 2015;457(3):262–6.
Fiorillo C, Astrea G, Savarese M, Cassandrini D, Brisca G, Trucco F, Pedemonte M, Trovato R, Ruggiero L, Vercelli L. MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of italian patients. Orphanet J Rare Dis. 2016;11(1):1–14.
Lamont PJ, Wallefeld W, Hilton-Jones D, Udd B, Argov Z, Barboi AC, Bonneman C, Boycott KM, Bushby K, Connolly AM. Novel mutations widen the phenotypic spectrum of slow skeletal/β‐cardiac myosin (MYH 7) distal myopathy. Hum Mutat. 2014;35(7):868–79.
Tasca G, Ricci E, Penttilä S, Monforte M, Giglio V, Ottaviani P, Camastra G, Silvestri G, Udd B. New phenotype and pathology features in MYH7-related distal myopathy. Neuromuscul Disord. 2012;22(7):640–7.
Ama P, Simoneau J, Boulay M, Serresse O, Theriault G, Bouchard C. Skeletal muscle characteristics in sedentary black and caucasian males. J Appl Physiol. 1986;61(5):1758–61.
Simoneau JA, Bouchard C. Genetic determinism of fiber type proportion in human skeletal muscle. FASEB J. 1995;9(11):1091–5.
Nier V, Schultz I, Brenner B, Forssmann W-G, Raida M. Variability in the ratio of mutant to wildtype myosin heavy chain present in the soleus muscle of patients with familial hypertrophic cardiomyopathy. A new approach for the quantification of mutant to wildtype protein. FEBS Lett. 1999;461(3):246–52.
Viswanathan MC, Tham RC, Kronert WA, Sarsoza F, Trujillo AS, Cammarato A, Bernstein SI. Myosin storage myopathy mutations yield defective myosin filament assembly in vitro and disrupted myofibrillar structure and function in vivo. Hum Mol Genet. 2017;26(24):4799–813.
Li L-l, Li R, Liu D. The association of the Arg1277Gln mutation in the MYH7 gene with myosin storage myopathy in a chinese family. 2020.
Dahl-Halvarsson M, Pokrzywa M, Rauthan M, Pilon M, Tajsharghi H. Myosin storage myopathy in C. elegans and human cultured muscle cells. PLoS ONE. 2017;12(1):e0170613.
Acknowledgements
Our heartfelt gratitude goes to the family members, who kindly allowed the documentation of their story with the aim of enhancing collective knowledge regarding this condition. This research was conducted by Rajaie Cardiovascular Medical and Research Center, Tehran, Iran, and was approved by its ethics committee (IR.RHC.REC.1399.019).
Funding
The authors received no specific funding for this research.
Author information
Authors and Affiliations
Contributions
NN and NM drafted the work.SK and MM designed the project.SK performed WES.MP collected the data. MM and YN surveyed the patients clinically.All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Consent for publication
Not applicable.
Ethics approval and consent to participate
The present study was performed in accordance with the Declaration of Helsinki and approved by Rajaie Cardiovascular Medical and Research Center (approval number: IR.RHC.REC.1399.019). Written informed consent was obtained from all participants for their participation and publication of this report..
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Naderi, N., Mohsen-Pour, N., Nilipour, Y. et al. A novel heterozygous missense MYH7 mutation potentially causes an autosomal dominant form of myosin storage myopathy with dilated cardiomyopathy. BMC Cardiovasc Disord 23, 487 (2023). https://doi.org/10.1186/s12872-023-03538-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-023-03538-8