
Abstract
Background
Although systemic lupus erythematosus (SLE) can affect the cardiovascular system in many ways with diverse presentations, a severe cardiogenic shock secondary to SLE myocarditis is infrequently described in the medical literature. Variable presenting features of SLE myocarditis can also make the diagnosis challenging. This case report will allow learners to consider SLE myocarditis in the differential and appreciate the diagnostic uncertainty.
Case presentation
A 20-year-old Filipino male presented with acute dyspnea, pleuritic chest pain, fevers, and diffuse rash after being diagnosed with SLE six months ago and treated with hydroxychloroquine. Labs were notable for leukopenia, non-nephrotic range proteinuria, elevated cardiac biomarkers, inflammatory markers, low complements, and serologies suggestive of active SLE. Broad-spectrum IV antibiotics and corticosteroids were initiated for sepsis and SLE activity. Blood cultures were positive for MSSA with likely skin source. An electrocardiogram showed diffuse ST-segment elevations without ischemic changes. CT chest demonstrated bilateral pleural and pericardial effusions with dense consolidations. Transthoracic and transesophageal echocardiogram demonstrated reduced left ventricular ejection fraction (LVEF) 45% with no valvular pathology suggestive of endocarditis. Although MSSA bacteremia resolved, the patient rapidly developed cardiopulmonary decline with a repeat echocardiogram demonstrating LVEF < 10%. A Cardiac MRI was a nondiagnostic study to elucidate an etiology of decompensation given inability to perform late gadolinium enhancement. Later, cardiac catheterization revealed normal cardiac output with non-obstructive coronary artery disease. As there was no clear etiology explaining his dramatic heart failure, endomyocardial biopsy was obtained demonstrating diffuse myofiber degeneration and inflammation. These pathological findings, in addition to skin biopsy demonstrating lichenoid dermatitis with a granular “full house” pattern was most consistent with SLE myocarditis. Furthermore, aggressive SLE-directed therapy demonstrated near full recovery of his heart failure.
Conclusion
Although myocarditis during SLE flare is a well-described cardiac manifestation, progression to cardiogenic shock is infrequent and fatal. As such, SLE myocarditis should be promptly considered. Given the heterogenous presentation of SLE, combination of serologic evaluation, advanced imaging, and myocardial biopsies can be helpful when diagnostic uncertainty exists. Our case highlights diagnostic methods and clinical course of a de novo presentation of cardiogenic shock from SLE myocarditis, then rapid improvement.
Similar content being viewed by others


Background
Systemic lupus erythematosus (SLE) is an autoimmune disorder with a variable presentation affecting multiple organ systems. Typical presenting SLE features include constitutional symptoms, cutaneous involvement, alopecia, oral ulcers, arthritis, renal involvement, and common cardiac symptoms, including chest pain, dyspnea, palpitations, and lower extremity swelling [1]. Cardiac manifestations include conduction disturbances, ischemia, cardiomyopathy, pericarditis, and myocarditis; however, rapid progression leading to cardiogenic shock is rare [2]. Clinical myocarditis occurs in nearly 5–10% of SLE patients, although post-mortem studies suggest a higher prevalence of subclinical myocarditis than previously known [3]. Viral infections are the most common cause of myocarditis in the United States and other developed countries; however, other causes include alcohol, drugs, radiation, and autoimmune diseases with SLE and granulomatosis with polyangiitis (GPA) being the two most common causes [4]. There is no definitive consensus for optimal treatment, but the combination of immunosuppressive therapy and medical treatment of heart failure has shown favorable outcomes. Here we describe the clinical course of a 20-year-old male with a de novo presentation of cardiogenic shock secondary to SLE myocarditis with the diagnostic challenges we encountered.
Case presentation
Our patient is a 20-year-old Filipino male diagnosed with SLE 6 months prior and was started on hydroxychloroquine. The patient presented to the emergency department after sudden onset of dyspnea, pleuritic chest pain, generalized fatigue, dry cough, fevers and chronic progressive rash. His review of systems was negative for sick contacts, recent travel, engagement in high-risk sexual activity, intravenous drug use, was a lifetime nonsmoker, and did not consume alcohol. On evaluation, vitals were notable for normal blood pressure (118/72 mmHg), tachycardia (118 beats per minute), tachypnea (respiratory rate 22 breaths per minute), and fever (100.8 °F). His cardiovascular exam was unremarkable for murmurs, elevated jugular venous pressure, splinter hemorrhages, or lower extremity edema. No cyanosis, clubbing, scleral icterus, organomegaly, or lymphadenopathy was appreciated. The skin examination showed no oral ulcers or alopecia, but demonstrated hyperpigmented to violaceous, scaly plaques with excoriated papules involving the bilateral extremities, back, and chest worsened as shown in Fig. 1. A prior skin punch biopsy of a similar rash (Fig. 2) showed lichenoid dermatitis with features of interface changes and chronic inflammation suggestive of cutaneous SLE. This rash had improved from his index presentation following treatment with hydroxychloroquine, though not fully resolved. Laboratory data on admission revealed the following: leukopenia, anemia, and non-nephrotic range proteinuria without active urinary sediment on urine microscopy. There was evidence of acute myocardial injury with elevated cardiac biomarkers along with an elevated N-terminal pro-brain natriuretic peptide. Additionally, inflammatory biomarkers were elevated with hypocomplementemia, positive antinuclear antibody (ANA) with anti-Smith, ribonucleoprotein, chromatin, SS-A, double-stranded DNA, perinuclear anti-neutrophil cytoplasmic antibodies (pANCA), and myeloperoxidase positivity. Comprehensive laboratory evaluation for additional workup and the reference ranges is shown in Table 1. His electrocardiogram and telemetry monitoring demonstrated normal sinus rhythm with diffuse < 1 mm ST-segment elevations in leads without any ischemic changes. Initial chest x-ray showed no acute cardiopulmonary process. Further infectious evaluation demonstrated positive blood cultures for methicillin-sensitive Staphylococcus aureus (MSSA) with a likely skin source from aggressively scratching a purulent lesion on the right anterior thigh. Initial transthoracic echocardiogram showed normal LVEF with all other normal parameters. Given concerns for sepsis secondary to presumed cellulitis, in the setting of SLE flare, he was treated with broad spectrum intravenous (IV) antibiotics and corticosteroids (methylprednisolone 1 g/kg for three days) before his transfer to our institution. Given concerns for endocarditis with persistent fevers on arrival, repeat transthoracic and transesophageal echocardiograms demonstrated mildly reduced LVEF 45% with no valvular pathology. MSSA bacteremia resolved within 72 h of intravenous antibiotics (cefazolin 2 g IV every 8 h), but the patient later developed a rapid cardiopulmonary decline on hospital day #5 with worsening dyspnea, pleuritic chest pain, and hemoptysis. A repeat chest x-ray (Fig. 3) and CT chest (Fig. 4) were obtained demonstrating dense pulmonary bilateral pleural effusions with dense consolidations, pulmonary nodules, possible pulmonary emboli within the left subsegmental pulmonary artery branches, and pericardial effusions. A repeat transthoracic echocardiogram demonstrated a severely reduced LVEF 25%, moderate global hypokinesis, and small pericardial effusion without evidence of tamponade (Fig. 5). Other abnormal findings included elevated right atrial pressures (15 mmHg) and mild pulmonary hypertension (43 mmHg) with a normal left atrial pressure, right ventricular systolic function, left ventricular diastolic function, and valvular function. Given clearance of bacteremia with IV antibiotics, cardiogenic shock was favored over septic shock. Intravenous vasopressors (norepinephrine, 0.4mcg/kg/min IV infusion) for shock, diuretics, amiodarone drip for atrial fibrillation with a rapid ventricular response, heparin drip for submassive pulmonary emboli, high-dose corticosteroids (solumedrol 500 mg IV every 12 h) and hydroxychloroquine 400 mg daily for SLE related co-activity were initiated. A cardiac MRI (CMR) was subsequently pursued to clarify the cause of myocarditis, which demonstrated a LVEF of 25% and moderate global hypokinesis with worsening pericardial effusions (Fig. 6). Unfortunately, the patient’s poor respiratory reserve and tachycardia limited our ability to perform late gadolinium enhancement. With his continued clinical decline potentially requiring escalating hemodynamic support, he was transferred to a medical center with advanced heart failure treatment capabilities.

Skin examination on hospital day #1 revealing a diffuse hyperpigmented to violaceous rash with excoriated papules with overlying hemorrhagic crust and scales (white arrows), some coalescing into small plaques distributed on trunk, bilateral thigs, knees, anterior lower legs, and feet

Skin examination during an outpatient clinic visit 6 months prior to his admission revealing a diffuse hyperpigmented rash with excoriated papules coalescing into plaques (black arrows) involving his bilateral upper and lower extremities and chest

A portable chest x-ray demonstrating bilateral interstitial infiltrates with dense consolidations in lung bases, bilateral pleural effusions, and pericardial effusion

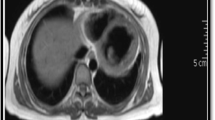
An axial view on computed tomography (CT) chest imaging obtained after the patient’s rapid cardiopulmonary decline demonstrating dense pulmonary consolidations, diffuse pulmonary nodules, and moderate pericardial (black arrow) and bilateral pleural effusions white arrows)

An apical four-chamber view on transthoracic echocardiogram obtained on the day of the patient’s cardiopulmonary decline (HD #6) demonstrating a moderate-sized pericardial effusion (white arrows)

A Cardiac MRI, T-2 weighted axial image of diastolic phase with late gadolinium enhancement sequence, demonstrating left ventricular dilation and large bilateral pleural effusions and pericardial effusions (white arrows). Myocardial tissue characterization with late gadolinium hyperenhancement was unable to be performed due to patient’s inability to lie recumbent and tachypnea
The differential etiologies leading to a precipitous cardiopulmonary decline in our SLE patient included infectious causes, autoimmune causes including an overlapping ANCA vasculitis, ischemic etiologies, septic cardiomyopathy, metabolic, and other toxic causes such as hydroxychloroquine-induced cardiomyopathy. An extensive infectious work-up included negative viral and fungal serologies, negative surveillance blood cultures, and respiratory cultures as listed in Table 1. The lack of arthritis, thrombocytopenia, and alveolar hemorrhage with ongoing systemic features better fit SLE, than overlapping ANCA vasculitis. In the absence of any other etiology to explain such a dramatic decline, a diagnosis of SLE myocarditis was most likely. Echocardiogram was repeated on hospital day #7 at the outside institution, which showed interval worsening of LVEF 10% with global hypokinesis, moderate concentric hypertrophy (LV mass index: 143.2 g/m2), and moderate pericardial effusion. Other echocardiogram parameters were normal. In addition to aforementioned therapy for SLE, medical management of his heart failure was instituted with angiotensin receptor neprilysin inhibitor (sacubitril/valsartan 24–26 mg twice daily), b-blocker (carvedilol 6.25 mg twice daily), digoxin 0.125 mg daily, amiodarone 200 mg daily, and mineralocorticoid receptor antagonist (spironolactone 12.5 mg daily). Following a relative improvement in hemodynamics, an endomyocardial biopsy was performed on hospital day #10 to elucidate the etiology of decompensation. Concurrently, a right and left heart cardiac catheterization and coronary angiogram were performed for a comprehensive cardiac workup, which revealed normal right and left-sided filling pressures, no evidence of pulmonary hypertension, normal cardiac output and index, and non-obstructive coronary artery disease. Later, his endomyocardial biopsy demonstrated extensive myocardial degeneration with architectural disarray, neoangiogenesis, and perivascular inflammation (Figs. 7, 8) suggestive of myocarditis. Notably, there were no granulomas, giant cells, or cytoplasmic vacuolization. Additionally, a repeat skin biopsy of a thigh lesion performed earlier revealed lichenoid interface dermatitis with vacuolization, and chronic inflammation (Fig. 9) with diffuse immunofluorescence demonstrated a granular deposition of IgA, IgG, C3, and fibrin (full house pattern) at the dermoepidermal junction. These pathological results of skin and endomyocardial biopsies further supported systemic SLE. The patient rapidly improved as his echocardiogram on the day of discharge (hospital day #12) showed improved LVEF 45% with improving pericardial effusion and normal left ventricular mass, and wall thickness. He was weaned off vasopressors and discharged on medical therapy for heart failure, oral prednisone 20 mg daily, hydroxychloroquine 400 mg daily, trimethoprim-sulfamethoxazole double-strength 800/160 mg twice daily for cellulitis, and cefazolin 2 mg IV daily MSSA bacteremia.

Histology of endomyocardial biopsy from the right ventricle with hematoxylin and eosin stain depicting normal myocardium with interstitial edema (black arrows) with early fibrosis highlighted by trichrome stain (white arrows)

Histology of endomyocardial biopsy with hematoxylin and eosin stain depicting an abnormal myocardium region with diffuse myofiber degeneration with significant architectural disarray, extensive infiltration of macrophages (CD68 positive) admixed with a few lymphocytes (CD45 immunostaining), neoangiogenesis (black arrows), and perivascular inflammation. Specifically, no granulomas, giant cells, or cytoplasmic vacuolization are visualized here. The spectrum of morphologic findings was highly suggestive of SLE myocarditis

A histology section of a skin punch biopsy with hematoxylin and eosin stain demonstrating superficial lymphocytic and neutrophilic infiltration within epidermis and dermis, vacuolization of the basal cell layer and rare apoptotic keratinocytes (white arrow) seen in lichenoid (interface) dermatitis compatible with cutaneous lupus erythematosus
At one month follow-up in rheumatology clinic, the patient was started on additional steroid-sparing therapy with monthly infusions of cyclophosphamide (750 mg/m2) given systemic SLE with cutaneous manifestations. After completing four cycles of cyclophosphamide and steroid taper, cardiac MRI was repeated after 3 months to assess interval activity. His severely impaired LVEF and pericardial effusions were resolved with no evidence of myocardial scarring or evidence suggesting active SLE on LGE and edema sequences. Echocardiogram after 6 months also showed normal LVEF > 55%, left ventricular diastolic function and mass, wall motion, and global longitudinal strain similar to his baseline findings. Laboratory evaluation showed a significant improvement in his inflammatory markers and cytopenias (leukopenia and anemia) shown in Table 2. His SLE is currently treated with mycophenolate mofetil 100 mg bid (transitioned from cyclophosphamide given favorable side effect profile with potentially long-term therapy) for maintenance therapy with a prednisone taper (60 mg daily for three weeks, taper by 10 mg every two weeks down to 10 mg daily), hydroxychloroquine 400 mg daily, and aforementioned heart failure guideline-directed medical therapy. His functional status is classified as New York Heart Association (NYHA) class I with continued clinical recovery (Table 3).
Discussion
Systemic lupus erythematosus is an independent risk factor for cardiovascular disease with a tenfold increased risk of complications as cardiac involvement may indicate disease severity [5]. Any cardiac structural components can be affected and may manifest as myocarditis, pericarditis, noninfectious (Liebman–Sacks) endocarditis, vasculitis, myocardial infarction, and heart failure [5, 6]. The underlying chronic inflammatory state of SLE inciting an immunologic injury from immune-complex deposition and complement activation has been postulated as the underlying mechanism of pathogenesis for increased atherogenesis, autoantibody production, endothelial dysfunction noted on postmortem myocardial biopsies [7]. We suspect a similar immunologic mechanism that contributed to ramped disease course leading to a dramatic cardiopulmonary decline on our patient. The prevalence of clinical SLE Myocarditis is estimated to be 5–10%, although recent studies suggest a higher prevalence of subclinical myocarditis as supported by evidence of acute myocardial injury in 37% of SLE patients from a recent study [3]. Although many studies suggest an increased risk of mortality for any clinically active SLE myocarditis, the prognostic implications of subclinical myocardial injury are unknown [3]. Despite generally favorable outcomes in SLE myocarditis, severe cardiac impairment on index presentation is associated with significant morbidity and mortality [1] (Table 4).
The diagnosis of SLE is established according to the 2019 classification criteria of European League Against Rheumatism (EULAR) and American College of Rheumatologic (ACR) [1]. Although heterogenous presentation of SLE myocarditis can pose diagnostic challenges. Our patient was initially diagnosed with SLE according to the EULAR/ACR classification (positive ANA, positive anti-Smith/RNP/dsDNA/chromatin/Ro/Ribosomal-P antibodies, low complements, leukopenia, positive agglutinin test without hemolysis, alopecia, photosensitivity, malar rash, and arthritis) and concordant findings of skin biopsy suggestive of cutaneous SLE. On admission, the laboratory evaluation suggested ramped SLE activity, though it was uncertain whether it was the primary driver underlying his cardiopulmonary decompensation in the context of concurrent sepsis. When a clinical dilemma exists, a combination of serologic evaluation and advanced cardiac imaging modalities such as conventional echocardiography and cardiac magnetic resonance imaging can be helpful in the diagnostic evaluation. Increased gadolinium enhancement on CMR can help characterize the spectrum of inflammatory changes within the myocardium and speckle tracking features within echocardiography provide prognostic implications in evaluating both clinical and subclinical myocarditis [8, 9]. Unfortunately, CMR feature was precluded by the patient’s poor respiratory effort and tachycardia during the initial study. However, repeat CMR obtained at 6-months to evaluate for any residual effects of SLE myocarditis showed no evidence of myocardial scarring or SLE activity on LGE and edema sequences. Endomyocardial biopsy is the gold standard test when diagnostic uncertainly exists in ruling out other alternative causes; however, it’s not routinely performed given its potential for procedural-related risks and sampling error that stems from patchy tissue involvement leading non-diagnostic results [7, 10, 13,14,15]. After a nondiagnostic CMR, endomyocardial biopsy was pursued which showed typical histopathologic features of myocarditis. These include regions of architectural disarray from infiltration of inflammatory cells and hypereosinophilic fibers leading to myocardial edema and necrosis as depicted in our patient’s endomyocardial biopsy shown in Figs. 7, 8 [7, 11].
Differentiating between the etiologies of myocarditis can be a clinical dilemma, as evident in our patient. Other etiologies, including metabolic, toxic, ischemic, infectious, and other autoimmune causes, were excluded by a negative laboratory workup and nonobstructive coronaries on coronary angiography. Septic cardiomyopathy was initially considered with MSSA bacteremia and initial concerns for potential septic emboli and developing abscesses on CT imaging, although deemed less likely given negative respiratory and surveillance blood cultures from treatment with IV antibiotics, lack of persistent evidence on serial imaging, and clinical improvement with SLE-targeted therapy [4]. Further unremarkable infectious workup included viral serologies for respiratory viruses, hepatitis panel, HIV, EBV, CMV, and fungal serologies for coccidioidomycosis, blastomycosis, cryptococcus, and histoplasmosis. Positive CMV IgM serology was less likely to represent an invasive CMV disease as large pentamers of IgM frequently cause false positives on ELISA testing. Hydroxychloroquine cardiomyopathy was not contributory given the absence of cytoplasmic vacuolization on cardiac biopsy and short duration of exposure to hydroxychloroquine, which typically occurs after years [12]. In the context of positive p-ANCA and MPO, an overlap of ANCA-associated vasculitis with SLE was considered, but less likely in the absence of any other systemic overlapping features, which typically include arthritis, renal injury with active urinary sediment, thrombocytopenia, or alveolar hemorrhage [1]. Drug-induced myocarditis was less likely in the absence of exposure to commonly-associated drugs such as hydralazine, procainamide, sulfa, isoniazid, and illicit substances such as cocaine [1]. Results of an extensive diagnostic evaluation featuring serologic profile (positive ANA, anti-dsDNA), radiographic evidence of serositis (pleural and pericardial effusions), and clinical presentation (chest pain, constitutional symptoms, and rash), and dramatic improvement with immunosuppressive therapy suggested SLE be the culprit of cardiogenic shock in our patient.
There is a paucity of data on optimal treatment strategies for cardiogenic shock secondary to SLE myocarditis as current guidance is based upon consensus driven by case studies and observational data. Immunosuppression, usually with high-dose steroids is the cornerstone of treatment, although cyclophosphamide, azathioprine, mycophenolate, and intravenous immunoglobulin can be alternative options to control disease activity based on review of recent case reports described in literature [2, 3]. Cyclophosphamide was utilized as an agent for steroid-sparing therapy given that it has been commonly used in SLE nephritis, severe cutaneous SLE, and other reported cases of myocarditis due to its favorable side effect profile and his Asian ethnicity [3]. Treatment in refractory cases involves disease-modifying novel biologic agents such as canakinumab, belimumab, and rituximab have also shown a trend towards positive clinical outcomes [6]. Despite early data suggesting a trend towards positive outcomes with immunosuppressive therapy, further studies aimed to investigate definitive treatment are needed for optimal management of SLE myocarditis. The treatment of severe cardiomyopathy involves guideline-directed medical therapy (ACE or ARB or ARNI, BB, MRA, isosorbide dinitrate-hydralazine, SGLT-2 inhibitors) for heart failure; however, intractable cases may involve utilization of cardiac resynchronization therapy (CRT), intraaortic balloon pump (IABP), mechanical circulatory support device (MCS), extracorporeal membrane oxygenation (ECMO), to augment left ventricular function with cardiac transplantation reserved as a last resort [6]. Fortunately, our patient’s severely compromised cardiac function rapidly improved with the combination of immunosuppressive therapy and medical therapy for heart failure sparing utilization of advanced mechanical support (Fig. 10).

A line graph depicting the patient’s LVEF during his clinical course
Conclusion
Despite its low prevalence, SLE myocarditis should be promptly considered in SLE patients presenting with new-onset heart failure given high morbidity and mortality rates. Early diagnosis and treatment with immunosuppressive therapy and heart failure medical management may lead to positive clinical outcomes. This case highlights a rare index cardiac presentation of SLE myocarditis leading to cardiogenic shock with rapid clinical recovery on immunosuppressive therapy and supportive care.
Availability of data and materials
Not applicable.
Abbreviations
- SLE:
-
Systemic lupus erythematosus
- ANA:
-
Antinuclear antibody
- RNP:
-
Ribonucleoprotein
- ENA:
-
Extractable nuclear antigen antibodies panel
- dsDNA:
-
Double stranded DNA
- GPA:
-
Granulomatosis with polyangiitis
- pANCA:
-
Perinuclear anti-neutrophil cytoplasmic antibodies
- MPO:
-
Myeloperoxidase
- MSSA:
-
Methicillin-sensitive Staphylococcus aureus
- PR-3:
-
Proteinase 3
- LVEF:
-
Left ventricular ejection fraction
- NT pro-BNP:
-
N-terminal pro-brain natriuretic peptide
- NYHA:
-
New York Heart Association classification of heart failure
References
Aringer M, Johnson SR. Classifying and diagnosing systemic lupus erythematosus in the 21st century. Rheumatology (Oxford). 2020;59(5):4–11.
Baquero G, Banchs J, Naccarelli G, Gonzalez M, Wolbrette D. Cardiogenic shock as the initial presentation of systemic lupus erythematosus: a case report and review of the literature. Congest Heart Fail. 2012;18(6):337–41.
Neonaki A, Filiopoulos V, Bonou M, Boletis IΝ, Cholongitas E. Acute liver injury as initial manifestation of systemic lupus erythematosus-induced myocarditis. Rheumatol Int. 2021.
Lin H, Wang W, Lee M, Meng Q, Ren H. Current status of septic cardiomyopathy: basic science and clinical progress. Front Pharmacol. 2020;211:10.
Kreps A, Paltoo K, McFarlane I. Cardiac manifestations in systemic lupus erythematosus: a case report and review of the literature. Am J Med Case Rep. 2018;6(9):180–3.
Durrance RJ, Movahedian M, Haile W, Teller K, Pinsker R. Systemic Lupus Erythematosus Presenting as Myopericarditis with Acute Heart Failure: A Case Report and Literature Review. Case Rep Rheumatol. 2019.
Arbustini E, Gavazzi A, Dal Bello B, Morbini P, Campana C, Diegoli M, Grasso M, Fasani R, Banchieri N, Porcu E, Pilotto A, Ponzetta M, Bellini O, Lucreziotti S, Viganò M. Ten-year experience with endomyocardial biopsy in myocarditis presenting with congestive heart failure: frequency, pathologic characteristics, treatment and follow-up. G Ital Cardiol. 1997;27(3):209–23.
Du Toit R, Herbst PG, Ackerman C, Pecoraro AJ, Claassen D, Cyster HP, Reuter H, Doubell AF. Outcome of clinical and subclinical myocardial injury in systemic lupus erythematosus—a prospective cohort study. Lupus. 2020;30(2):256–68.
Mawlana W, Tolba O, Abdel Nabi H, Shabana A. Differentiation of bilayer interventricular septum function in children with systemic lupus erythematosus by tissue Doppler and speckle tracking imaging. Echocardiography. 2021;38(1):16–24.
Joyce E, Fabre A, Mahon N. Hydroxychloroquine cardiotoxicity presenting as a rapidly evolving biventricular cardiomyopathy: key diagnostic features and literature review. Eur Heart J Acute Cardiovasc Care. 2013;2(1):77–83.
Noutsias M, Seeberg B, Schultheiss HP, Kühl U. Expression of cell adhesion molecules in dilated cardiomyopathy: evidence for endothelial activation in inflammatory cardiomyopathy. Circulation. 1999;99(16):2124–31.
Jarrot P-A, et al. Systemic lupus erythematosus and antineutrophil cytoplasmic antibody-associated vasculitis overlap syndrome in patients with biopsy-proven glomerulonephritis. Medicine. 2016;95:22.
Katzmann JL, Schlattmann P, Rigopoulos AG, Noutsias E, Bigalke B, Pauschinger M, Tschope C, Sedding D, Schulze PC, Noutsias M. Meta-analysis on the immunohistological detection of inflammatory cardiomyopathy in endomyocardial biopsies. Heart Fail Rev. 2020;25(2):277–94.
Lazzarotto T, Dalla Casa B, Campisi B, Landini MP. Enzyme-linked immunoadsorbent assay for the detection of cytomegalovirus-IgM: comparison between eight commercial kits, immunofluorescence, and immunoblotting. J Clin Lab Anal. 1992;6(4):216–8.
Zhang L, Zhu YL, Li MT, Gao N, You X, Wu QJ, Su JM, Shen M, Zhao LD, Liu JJ, Zhang FC, Zhao Y, Zeng XF. Lupus myocarditis: a case-control study from China. Chin Med J (Engl). 2015;128(19):2588–94.
Acknowledgements
Not applicable.
Funding
There is no source of funding or any form of financial assistance that contributed to this case report.
Author information
Authors and Affiliations
Contributions
All authors have read and approved the final manuscript. JJR (*)—Internal medicine resident who compiled the data on patient’s extensive hospital course, incorporated background information and relevant evidence available to date that’s applicable to the context of this topic. JJR is also the primary author of this manuscript. CRR, JH, and JJW assisted in reviewing and editing the final manuscript.
Author information
Jaydeep J. Raval: is a PGY-3, Internal Medicine resident at the Naval Medical Center San Diego with future interests in pursuing cardiology. Christina Rodriguez Ruiz: is currently a Cardiology fellow at Scripps Cardiology Fellowship program. James Heywood: currently serves as a faculty member specializing in heart failure at Scripps Hospital. Jason J. Weiner: is currently a staff rheumatologist at the Naval Medical Center San Diego and serves as one of the associate program directors in the Internal Medicine Residency.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written consent for the identifying images, clinical details, and radiographic and histological images was obtained from the patient involved in our case report. A proof of consent to publish is available upon request.
Competing interests
The authors declare that they have no competing interests. The views expressed in this article reflect the results of research conducted by the author and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the United States Government.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Raval, J.J., Ruiz, C.R., Heywood, J. et al. SLE strikes the heart! A rare presentation of SLE myocarditis presenting as cardiogenic shock. BMC Cardiovasc Disord 21, 294 (2021). https://doi.org/10.1186/s12872-021-02102-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-021-02102-6