
Abstract
Background
The transposons of the hAT superfamily are the most widespread transposons ever known. SLEEPER genes encode domesticated transposases from the hAT superfamily, which may have lost their transposable functions during long-term evolution and transformed into host proteins that regulate plant growth and development.
Results
This study identified 162 members of the SLEEPER gene family from Brassica napus. These members are widely distributed on 19 chromosomes, mainly in the Cn subgenome, and have promoters with various cis-acting elements related to hormone regulation, abiotic stress, and growth and development regulation. Most of the genes in this family contain similar conserved domains and motifs, and the closer the genes are distributed on evolutionary branches, the more similar their structures are. Transcriptome sequencing performed on tissues at different growth stages from B. napus line 3529 indicated that these genes had different expression patterns, and nearly half of the genes were not detectably expressed in all samples.
Conclusions
This study investigated the gene structure, expression patterns, evolutionary features, and gene localization of the SLEEPER family members to confirm the significance of these genes in the growth of B. napus, providing a reference for the study of transposon domestication and outstanding genetic resources for the genetic improvement of B. napus.
Similar content being viewed by others


Background
Transposable elements (TEs) are DNA segments that can move from one locus to another in the host genome. Once thought to be “junk DNA” of no use to the organism [1], TEs have been found to account for a dramatic proportion of the genomes. For example, at least 10% of the genome of the flowering plant Arabidopsis thaliana [2], about 35% in rice [3], and up to 85% of the maize genome [4], which suggested that TEs play a very important role in genome expansion and gene regulation of organisms with large genomes.
Based on their transposition mechanism, TEs are classified into three types: Class I transposons (retrotransposons), Class II transposons, and Helitrons. Class I transposons have a transposition mechanism called “copy-and-paste” that uses DNA as a template, transcribes it into mRNA, then reverse transcribes the mRNA into cDNA, and integrates the cDNA into a new site in the genome [5]. Therefore, Class I transposons are also known as RNA-type transposons [6]. Class II transposons use a DNA-mediated mechanism called “cut-and-paste” that does not involve reverse transcription of RNA. In this process, a Class II transposon dissociates from its original position and re-integrates into the chromosome under the action of transposase, and the chromosome broken in the original position is repaired by the DNA repair mechanism. Therefore, Class II transposons are also called DNA-type transposons [7]. Helitrons is a new type of transposon identified in the A. thaliana genome by algorithms based on repetitive sequences in recent years. Unlike retrotransposons and transposons, Helitrons uses a rolling circle mechanism for transposition. During the rolling circle replication process, gene fragments can be captured and carried, which also helps facilitate genome evolution [8].
It has been shown that during domestication, transposons, like functional genes, were retained and transformed into host proteins, such as chromatin-associated proteins and transcription factors, or were gradually lost in the evolutionary process called transposon domestication [9]. Transposons are no longer perceived as non-functional or even harmful genes. During evolution, the insertion of transposons may cause new mutations that affect the regulatory regions in which they are located and transform these transposons into new genes that never existed before [10]. This process is called molecular domestication, and these new genes are called transposon-derived genes or domesticated genes, which play an important role in the growth and development of organisms [11]. Over the years, many important transcription factors have been identified as a result of transposon domestication through reverse transcription events, such as Far1/Fhy3 (far-red impaired response protein 1/far-red elongated hypocotyl 3) in plants, which is derived from the MULE-type DNA transposon and is involved in the far-red light response of plants after being domesticated [12], the recombination-activating gene products RAG1/2 protein in vertebrates, which is a key element of the vertebrate adaptive immune system and is essential in the development of the complex placenta [13]; the BEX/TCEAL cluster identified in mammals, which is derived from LINE-type superfamily transposase and plays a crucial role in metabolic pathways related to proliferation and differentiation in the nervous system [14]; and PANDA, a rice Harbinger transposon-derived gene, could epigenetically coordinate panicle and grain size in Oryza sativa [15].
The hAT (hobo, Activator, and Tam3) superfamily from Class II transposons, one of the most widespread families of known transposable elements [16], is common in plants, fungi, animals, and humans [17]. The SLEEPERs genes that encode a domesticated transposase from the hAT superfamily are structurally conserved in most species and usually consist of a C2H2 type BED-zinc finger domain (Zf-BED) and a hAT dimerization domain [18]. The Zf-BED has a function to bind DNA and is derived from a transposable element [19]. The hAT dimerization domain was first identified in the maize Ac transposase and was thought to be present at the C-terminus of transposases from the hAT family [20]. It has been shown in vivo and in vitro that loss of the dimerization structure is directly related to loss of transposase activity, and the hAT dimerization domain is relatively more conserved [21]. All known SLEEPERs originate from DAYSLEEPER in A. thaliana and are only expressed in plants [22]. Either deletion or overexpression of these genes can cause a dramatic phenotype in A. thaliana. The mutant seedlings grew slowly, without enlargement of cotyledon or normal development of leaves and floral organs. In terms of molecular function, DAYSLEEPER binds not only to proteins but also to DNA, and has protein dimerization activity [23]. It is suggested that DAYSLEEPER may have been domesticated from a group of hAT transposons to transcription factors that play a crucial role in growth and development regulation of A. thaliana instead of being transposable [24]. However, in other species, more detailed classification and function studies of DAYSLEEPER are scarce.
Brassica napus (AACC = 38) is an allotetraploid generated by natural hybridization between two diploid species, Brassica rapa (AA = 20) and Brassica oleracea (CC = 18). The proportion of TEs in the genome varies depending on the cultivars of B. napus, with up to 61.83% (569.75 Mb) of TEs in the ZS11_PB genome [25]. In this study, we identified the SLEEPER family members from the whole genome of B. napus by bioinformatics methods. Their genetic relationships, molecular structural features and expression patterns in different tissues at different developmental stages were then analyzed. We also identified the subcellular localization of some of these members.
Results
Identification of SLEEPER genes in B. napus
In this study, candidate proteins were identified from B. napus genome protein sequences (GCF_00686985.2) using HMM file of the conserved domain. The results of domain-based extraction and bidirectional BLAST optimal comparison screening were summarized and duplicate values were removed. Eventually, 162 SLEEPER family members were identified (Table S1).
The lengths of these SLEEPER proteins range from 102 to 1908. The molecular weights (MW) vary from 11,422 to 215,793, and the theoretical isoelectric points (Theoretical_pI) of SLEEPER proteins range from 4.44 to 9.54 (Table S2).
Chromosomal location and phylogenic analysis of SLEEPERs in B. napus
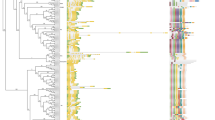
To understand the evolutionary relationship of SLEEPERs from B. napus, we constructed an ML tree among 342 SLEEPER proteins, including 162 in B. napus, 93 in B. oleracea, 51 in B. rapa, 2 in (A) thaliana and 34 in O. sativa. According to Knip’s naming convention of SLEEPER genes in plants, such as RICESLEEPER or VINESLEEPER [18], and considering (B) napus, also known as rapeseed, we named some BnSLEEPERs genes as RAPESLEEPER. The distance of the branches of the ML tree showed that all the BnSLEEPERs could be classified into 5 clusters (Fig. 1). Among them, BnRAPESLEEPER1 (BnRAPE1.1-1.2) only contained two members, which did not cluster with other SLEEPERs on the branches of the tree but still had the features of SLEEPER proteins. BnDAYSLEEPER (BnDAYSLEEPER1-31), which contained 31 members, had higher homology to AtDAYSLEEPER than the others did. The rest of the family members were divided into BnRAPESLEEPER2 (23 members, hereafter referred to as RAPE2.1-2.23.), BnRAPESLEEPER3 (49 members, hereafter referred to as RAPE3.1-3.49) and BnRAPESLEEPER4 (57 members, hereafter referred to as RAPE4.1-4.57), according to their position on the evolutionary branch (Fig. 1).

Phylogenetic analysis of SLEEPER genes in Arabidopsis thaliana, Brassica napus, Brassica rapa, Brassica oleracea, and Oryza sativa. (a) Phylogenetic tree trunk of 342 SLEEPER genes. It was constructed by maximum likelihood (ML) method based on multiple sequence alignment of 342 amino acid sequences of SLEEPER genes, different numbers indicated each node of the phylogenetic tree. (b) Branches on each node of the trunk of the phylogenetic tree
After localizing 162 SLEEPER genes on the chromosomes of B. napus, it was found that these SLEEPER genes were unevenly distributed on subgenome Cn and An chromosomes. Most of the family members were distributed on Cn, containing 95 members. 14 members that had not been successfully assembled to the chromosomes of B. napus due to the chromosomal complexity were distributed on unscafflold contigs. There were only 53 members localized on An. In terms of gene distribution on chromosomes, most members on An were located in regions where genes were densely distributed, while on Cn more SLEEPER members were located in regions of higher non-coding region density (Fig. 2).

Chromosomal location and duplication of 162 SLEEPER genes in B. napus. Different colors represent varying gene densities, with blue to red indicating low to high gene density
Gene structure, protein motif, conserved domains
The motifs, conserved domains and cis-acting elements were analyzed in order of their phylogenetic relationships. Most of the open reading frames (ORFs) were 500–3000 bp in length, with some of them exceeding 9000 bp even 20,000 bp, such as RAPE4.11 and RAPE4.20. CDSs of these 162 SLEEPER genes were 306–5727 bp in length, with RAPE1.1 having the longest CDS sequence and RAPE4.39 the shortest (Fig. 3b).
Protein motif and conserved domains analysis indicated that most members of the SLEEPER gene family each contained three conserved domains, Dimer_Tnp_hAT (Pfam05699), ZnF_BED (Cl02703), and a DUF4413 (Pfam14372). Notably, RAPE2 contained no DUF4413 domain, with DUF659 domain instead. Besides, some members of the family contained certain exclusive conserved domains. For instance, RAPE4.23 contained a RNase_H_like domain (PF04308), RAPE4.20 contained a PLN02308 domain that was classified as a model that may span more than one domain, RAPE4.5 contained a Zf_RVT domain (PF13966), and RAPE3.23 contained a U5 snRNP spliceosome subunit called PRP8 (COG5178). A few of the members contained only one or two of the three conserved domains. For example, RAPE4.56 and RAPE4.57 contained only Dimer_Tnp_hAT, RAPE4.32 and RAPE4.43 contained only ZnF_BED, and there was none of the three conserved domains in RAPE4.47 (Fig. 3a).
The synthesis of motif analysis and domain analysis indicated that Dimer_Tnp_hAT always contains motif6 and motif2, DUF4413 often contains motif4, and that motif1, motif3 and motif5 rarely overlap with any of the conserved domains (Fig. 3a).
It is noteworthy that almost all members of RAPE1 and RAPE2 contained conserved domains different from those of other family members, without any similar conserved motifs to other family members, particularly RAPE1.1, despite having the largest molecular weight among its proteins, contained none of the characterized domains for the SLEEPER gene family, namely Dimer_Tnp_hAT domain, DUF4413 domain and ZnF_BED domain, sharing the only similarity with other family members by containing motif2.

Gene structure, motif, conserved domain and cis-elements analysis of BnSLEEPERs. (a) Distribution of conserved domains and motif elements, the colored bars represent different motifs, and the dashed boxes in various colors represent the location of the conserved domains; (b) Gene structure, the green bars represent the UTRs, and the yellow bars represent the exons; (c) Cis-elements analysis of the B. napus SLEEPER genes family promoter regions 1500 bp upstream of the initial codons. Scale markers represent the length of genes (bp) or proteins (aa)
Cis-elements analysis of promoter regions
The upstream 1500 bp sequence of the gene initiation codon was selected to identify the enriched cis-acting elements (CREs), to further speculate on the possible pathways involved in the SLEEPER family of genes. After selecting the highly enriched CREs, the results showed that there were 21 types of cis-acting elements in the promoters of all SLEEPER family members, with the undefined cis-acting elements such as TATA-box and CAAT-box excluded. Based on the metabolic pathways and functions they participated in, we classified these cis-acting elements into three groups, i.e. plant hormones responsiveness elements, environmental stress responsiveness elements and development regulation elements. Among the plant hormones responsiveness elements were abscisic acid responsiveness elements, gibberellin responsiveness elements, salicylic acid responsiveness elements, auxin responsiveness elements and MeJA-responsiveness elements. In the environmental stress responsiveness elements were anaerobic induction elements, light responsiveness elements, low-temperature responsiveness elements, drought-inducibility elements, wound responsiveness elements and defense and stress responsiveness elements. And the development regulation elements included meristem expression elements, endosperm expression elements, root-specific elements, seed-specific regulation elements, differentiation of the palisade mesophyll cells elements, zein metabolism regulation elements, phytochrome down-regulation expression elements, circadian control elements, cell cycle regulation elements and flavonoid biosynthetic genes regulation elements (Fig. 3c).
Furthermore, all BnSLEEPERs contained environmental stress responsiveness elements, 152 genes could be associated with plant hormones responsiveness, and only 77 genes were involved with plant development regulation, indicating that the SLEEPER family is related to a variety of plant hormones and environmental stress responsiveness and that some members may participate in plant development regulation (Table S3). Therefore, SLEEPER family genes may play an important role in plant hormones metabolism and environmental stress responsiveness. Significantly, the fact that light response-related CREs were widely distributed among all SLEEPER family genes suggested that SLEEPER genes are probably critical in plant response to light.
Synteny and gene duplication of SLEEPERs genes in B. napus
To investigate the evolutionary history by syntenic gene analysis, we traced the orthologous gene pairs in B. napus, between B. rapa and B. napus, and between B. oleracea and B. napus, respectively. The synteny analysis within species shows that all family members except two genes from RAPE1 have internal linear relationships. In subgenome An, there are four collinear gene pairs, all from DAYSLEEPER. There are six collinear gene pairs in subgenome Cn, one from RAPE4, two from RAPE3, and three from DAYSLEEPER. A total of twelve collinear gene pairs were identified between the An and Cn, of which eleven pairs are in the DAYSLEEPER group and one pair is in the RAPE2 group (Fig. 4a).
Gene family expansion proceeds mainly through five ways: whole genome duplication or polyploidization, tandem duplication, segmental duplication, retrotransposition, and exon duplication and shuffling. Considering the chromosomal distribution of the SLEEPER family genes and the results of synteny analysis, we suggest that the expansion of the SLEEPER gene family is mainly due to whole genome duplication, segmental duplication, and tandem duplication. Allotetraploid B. napus evolved from the ancestral B. oleracea and B. rapa, both of which are relatively ancient polyploid plants that have long undergone large-scale chromosomal rearrangements. For instance, DAYSLEEPER13-14, 23–29, 23–24 and other collinear gene pairs distributed in An and Cn, respectively, are very likely to have undergone ploidy during the polyploidy process. On the other hand, a large number of SLEEPER family genes are closely aligned on the same chromosome in the genome, forming clusters of genes with similar sequences and functions, such as the gene pairs DAYSLEEPER19-20, 24–25, RAPE3.40-3.41, RAPE4.20-4.22, RAPE2.7-2.8, etc. Therefore, tandem duplication is another major cause of the expansion of SLEEPER family members (Fig. 4a).

Syntenic and ka/ks analysis. (a) Syntenic analysis between An and Cn subgenomes in B. napus. Green lines link the duplicate gene pairs from Cn, blue lines link the duplicate gene pairs from An, and the red lines link the duplicate gene pairs from both An and Cn. Different colors represent varying gene densities, with blue to red indicating low to high gene density. (b) Syntenic analysis of SLEEPERs in B. oleracea, B. napus and B. rapa. Duplicate gene pairs are highlighted with red connecting lines. (c) Ka, Ks and Ka/Ks analysis of duplicate gene pairs. The black dots represent the outlier samples. Significance analysis was conducted by independent-samples t-test and black asterisks denote significance. *p < 0.05; **p < 0.01. Duplicate gene pairs were calculated by TBtools
To investigate the evolutionary process of the SLEEPER gene family in cruciferous species, this study further analyzed the homologous relationship between B. napus and B. rapa and B. oleracea. Among the 162 members from B. napus, 37 genes could be homologous in either B. rapa or B. oleracea, of which 21 could be found homologous in both B. rapa and B. oleracea, 13 genes could be homologous in B. oleracea only, and 3 genes could be homologous in B. rapa only. This suggested that the gene family is more expanded in B. oleracea than in B. rapa and that members of this gene family in B. napus probably originated mainly from B. oleracea during the evolutionary process, which is consistent with the results of gene localization analysis (Fig. 4b).
The Ka/Ks of An and Cn subgenomes of B. napus ranged from 0.043 to 0.513, with an average of 0.284. The Ka/Ks of B. napus and B. rapa ranged from 0.037 to 1.430, with an average of 0.271, and only one Ka/Ks value of duplicate gene pair over 1.0. The Ka/Ks of B. napus and B. oleracea ranged from 0.063 to 2.020, with an average of 0.472, and five Ka/Ks values of homolog gene pairs over 1.0 (Fig. 4c, Table S4). Ka/Ks = 1 indicated neutral evolution, Ka/Ks < 1 indicated purifying evolution, and Ka/Ks > 1 indicated positive selection [26]. It is suggested that in cruciferous species, while a few SLEEPER genes are evolving rapidly, most of them are relatively conserved.
Expression analysis of BnSLEEPERs genes during growth and development in different tissues
It was previously suggested that AtDAYSLEEPER may be involved in various plant growth and development activities as a transposase. To investigate whether members of the SLEEPER gene family have similar functions in B. napus, we analyzed their transcriptional levels in different tissues at different growth stages in B. napus. These members all have multiple copies in allotetraploid B. napus, so the identification of the key members is important for a more in-depth study of the evolutionary process and relationship of SLEEPER members from transposons to transcription factors, which will help to identify excellent genetic resources for genetic improvement in B. napus.

Expression of 162 SLEEPER genes in different tissues at different stages. Heatmaps were displayed with log10 values for each gene. The color scale represents the relative expression levels from low (blue) to high (red). Seedling stage (ss), flowering stage (fs), pods/silique stage (ps)
The RNA-seq results showed that the expression of 74 SLEEPER genes were almost undetectable in all tissues at all stages, including 10 DAYSLEEPERs, 7 RAPE2s, 34 RAPE3s, and 23 RAPE4s (Fig. 5, Table S5). It is speculated that there may be two main reasons for this. One was the presence of retrotransposition during the evolutionary process, where the ancestral SLEEPER genes, as hAT transposons, lost their transposition function during long-term evolution and thus became transcription factors, and the retrotransposition happened during this process could form new genes that lack necessary regulatory sequences such as the UTR region, resulting in a large number of unexpressed pseudo genes. This hypothesis is supported by the results of gene structure analysis. Another reason is that we may have selected too few growth stages of B. napus and did not pick the stages or tissues in which these genes were expressed.

Expression analysis and subcellular location of 4 BnSLEEPERs. (a) The relative expression profile of BnDAYSLEEPER8, BnRAPESLEEPER2.12, 3.38 and 4.35 in 5 tissues of three different stages. Different letters above the bars indicated significantly different values (p < 0.05) calculated using one-way analysis of variance (ANOVA) followed by Tukey’s multiple range (b) The subcellular location of BnSLEEPERs above. Control1# was eGFP driven by 35 S promoter, and control2# was fusion protein of eGFP and BnNRAMP, confirmed membrane localized. Each box represents a length of 110 μm. (c) Correlation analysis between RNA-seq data and qRT-PCR data. Student’s t-test was used to calculate significance. The * and ** indicated P < 0.05 and P < 0.01 respectively
It was demonstrated in the TAIR database that AtDAYSLEEPER is mainly involved in the development of floral meristematic tissues and is essential for the development of structures such as ovules and carpels. We screened a total of 36 genes with FPKM values greater than 10 in any tissue or stage for individual analysis. Among them, RAPE2.2, 2.22, and 3.34 were mainly expressed from the flowering stage only, while RAPE4.44, and 4.9 had very low-level expression in roots and stems during the flowering stage (Fig. 5, Fig. S1). Furthermore, the expression levels of other members in BnSLEEPERs genes were not significantly tissue-specific, with relatively high-level expression in almost all stages and tissues. In conclusion, the whole SLEEPER gene family is involved in the regulation of the growth of various tissue in B. napus from seedling to silique stage. In addition, a correlation analysis of the qRT-PCR data and the FPKM results from the RNA-seq experiment showed that the expression of the four selected genes was closely correlated with each other (Fig. 6c). The range of R-value was between 0.69 and 0.94, using a significance level of p < 0.05 (Fig. S3).
To determine the roles that the SLEEPER gene family members played during plant growth and development, we constructed a SLEEPER protein interaction network in the model plant A. thaliana using the STRING database based on previous experiments and predicted interactions. Previous analysis with eight protein interactions predicted that AtDAYSLEEPER is most likely involved in chromatin remodeling, and most of the proteins predicted by experimental and bioinformatic approaches to interact with DAYSLEEPER by protein-protein interactions are chromatin remodeling proteins, such as ATRX, which participates in transcriptional regulation and chromatin remodeling through facilitating DNA replication in a variety of cellular environments and promoting the expression of chromatin remodeling factors PKL, PKR2 (involved in the repression of embryonic trait gene expression during and after seed germination) and CHR4 (regulating gene transcription through binding of histone and DNA) (Fig. S2).
Prediction and verification of subcellular location of BnSLEEPERs
To further explore the potential functions of domesticated SLEEPER genes, we performed signal peptide (SP) prediction, transmembrane helices (TH) analysis, and subcellular location prediction on these 162 members using various databases. The results showed that a total of 51 members were predicted to contain nuclear localization signal (NLS), 15 of them were predicted to be located in chloroplasts, and only two were predicted to contain signal peptides. In addition, among the 21 members predicted to contain TH, two contained NLS and were predicted to be localized in the chloroplast and nucleus (Table S6). Overall, most of the members were predicted to be localized in the nucleus or chloroplast, and a few were predicted to contain TH, while a very few might be localized in the Golgi apparatus, lysosomes, or even extracellularly.
To verify the subcellular localization of SLEEPERs genes, from the 36 highly expressed genes mentioned above (Fig. S1), we screened 4 genes according to the different predictions for tobacco transient transformation after fusing eGFP and then observed their localization. BnDAYSLEEPER8 with NLS (RRRRK) is predicted to be localized in the nucleus and not contain any SP or TH. BnRAPE3.38 is predicted to be located in the nucleus but contains no NLS, TH or SP. BnRAPE2.12 possesses both NLS ((KATKRKHR) and SP but lacks TH. BnRAPE4.35 only exclusively contains THP. It is worth mentioning that among the 36 genes, there are no genes with only SP, or genes with both SP and TH, or genes with both NLS and THP (Table S6).
The experimental results showed that BnDAYSLEEPER8 only located in the nucleus while BnRAPE3.38 was expressed not only in the nucleus but also in the cell membrane in small amounts. Although BnRAPE2.12 was predicted to contain SP and NLS but no TH, it was found in experiments to be expressed in chloroplasts, nucleus and cell membrane. The results showed that BnRAPE4.35 was also notably expressed in the cell membrane and appeared to be expressed in some unidentified intracellular regions excluding the nucleus even without SP (Fig. 6b). The experimental results indicated that the actual subcellular localization of some members did not exactly match the predicted results, and each member was expressed in the nucleus, suggesting that during transposon domestication, not all transposon-derived genes will be transformed into transcription factors, but possibly into other types of functional genes as well.
According to the RNA-seq and qRT-PCR data, these four genes relatively expressed highly in various tissues and stages in B. napus, except for RAPE4.35, the other three genes expressed highly in the pods. RAPE4.35 mainly expressed in the leaves and flowers, also relatively high in roots at flowering stage. As for RAPE3.38, the expression level of it in the leaves gets less and less, and on the contrary, the expression level in stems increased gradually. In addition, the same is true for DAYSLEEPER8. However, the overall change trend of RAPE2.12 expression was not obvious. (Fig. 6a, Fig. S2)
Discussion
Brassica napus is one of the most important oil crops in the world and is an ancient heterotetraploid species that evolved from the ancestral Brassica oleracea and Brassica rapa [27]. Among Brassica crops, B. oleracea and B. rapa diverged about 46 million years ago [28], and the natural hybridization of them that formed B. napus occurred about 7500 years ago [27]. During polyploidization events, a large number of duplicated chromosomal regions can be retained on the genome so that some genes may expand by segmental duplication [29], such as the CLE gene family in B. napus [30]. The results show that SLEEPER family genes are scarce in (A) thaliana, but abundant in (B) oleracea, B. rapa, or B. napus. The gene structure and gene expression patterns analysis suggested that there might be many pseudogenes in the SLEEPER gene family, which do not contain complete gene structures, missing important regions such as the UTR, and could not be detected to be expressed in the samples we selected. In terms of gene distribution on chromosomes, many homologous gene pairs are extremely close in physical distance, located on the same chromosome and less than 50 kb apart. The results above suggest that the SLEEPER family may have expanded rapidly in various ways leading to numerous members, such as genome polyploidization, segmental duplication, and tandem duplication. Furthermore, it was reported that the DALYSLEEPER gene was domesticated from the hAT transposon, which still contained the hAT-like transposable element, but lost sequences essential for transposition such as the terminal inverted repeat (TIR) [18]. Therefore, the uneven distribution of the SLEEPER gene family in the An and Cn subgenomes of the B. napus genome may be attributed to the fact that the Cn contains more transposons than the An, and that homologous exchanges were more frequent and active in the Cn than in the An during polyploidization [27]. This also suggested the possibility that retrotransposition replication is another major way for SLEEPER genes expansion.
Through bioinformatics analysis, this study provided a systematic interpretation of the molecular characteristics of the SLEEPER gene family, which is a very large and complex gene family in many cruciferous species. After identifying the genome-wide SLEEPER gene family in B. napus, we divided the members into five groups. The bioinformatics analysis showed that their gene structures and motifs are conserved, the types of cis-acting elements contained in their promoters are mostly consistent, and the functions of the members in different groups are not specific, suggesting that their gene functions may have become redundant during the evolutionary process. The results of transcriptional patterns in different growth stages and tissues of B. napus also confirmed this conjecture, with many genes having similar or even identical expression patterns, such as RAPE2.22 vs. RAPE2.2, DAYSLEEPER13 vs. RAPE2.10, and RAPE2.22 vs. RAPE2.19. Interestingly, almost all duplicated genes differ dramatically in their expression patterns, probably because these duplicated genes have undergone non-functionalization, neo-functionalization or sub-functionalization at the transcriptional level, thus evolving in different directions and taking on more functions [31].
The functions of SLEEPER genes in plants are poorly understood. Although the discoverers of DAYSLEEPER found that it is essential for plant growth and development, especially for meristem formation, the exact signal pathways and transcriptional regulation involved are still unknown [23]. In the human genome, the mammal-specific CCCH type BED-zinc finger domain proteins (ZBED transcription factors), which are homologous to the plant SLEEPER genes and domesticated from the hAT superfamily, have been shown to have important functions in the human body as transcription factors, although they also lack transposase activity but retain the zinc finger domain [32, 33]. For example, ZBED transcription factors have been verified to be involved in various cell development and immune response processes [34], such as regulating keratinocyte differentiation and being a marker of T cell failure in CD8 T cells [35].
In this study, the prediction and experiments of subcellular localization of SLEEPER members from different groups in B. napus showed that these proteins are mainly localized in the nucleus, with a small number of them expressed in the cytoplasm, and even some members are expressed in the cell membrane. Knip also demonstrated that AtDAYSLEEPER is localized in the nuclear and vesicular, suggesting that SLEEPERs genes may control gene expression with the ability of DNA binding as trans-acting elements and thus regulate plant growth and development processes. In addition, the DAYSLEEPER protein interaction network in (A) thaliana also provides an alternative hypothesis for the function of homologous genes in (B) napus, namely, SLEEPER genes containing zinc finger domains may regulate chromatin structure as part of the coactivator complex and contribute to the activation function of nuclear receptors and other factors. The above results can provide a concrete reference example for the domestication of “junk gene” transposase into a transcription factor with important functions, which is helpful for exploring the significance of transposons as direct selection targets in domestication, and for fully understanding the functions and roles of transposons in genome evolution. It also provides directions and clues for further genetic improvement breeding research of B. napus.
Conclusion
In this study, 162 members of the SLEEPER gene family were identified in B. napus by a combined method of BLAST and HMM search. The prediction of their structural and functional similarities and differences and the analysis of their promoter elements and protein subcellular localization were performed by molecular characterization and bioinformatic analysis of the entire gene family. With transcriptome analysis, their expression patterns in different tissues throughout the growth and development cycle of B. napus. Finally, considering the results of the identification of differential expression, co-expression network analysis and subcellular localization altogether, we further screened the core genes that could further explain the evolutionary process of ancient transposons losing their transposable function and then transforming into transcription factors or functional genes with an important role in plant growth and development. In conclusion, these genes will likely provide high-quality genetic resources for the genetic improvement of B. napus.
Materials and methods
Identification and classification of SLEEPER gene family members in B. napus and the other plants
Candidate SLEEPER gene family members of B. napus, B. rapa, B. oleracea, A. thaliana and O. sativa were retrieved from NCBI database (https://www/ncbi.nlm.nih.gov/genome/), the version of Genomic sequence and GFF Annotated feature files were Bra_napus_v2.0, CAAS_Brap_v3.01, BOL, TAIR10.1 and IRGSP-1.0, respectively.
Bidirectional blast alignment and Hidden Markov Model (HMM) search were carried out to obtain all the candidate gene family members. NCBI’s BLAST algorithm was used to perform alignments and homology analyses (e-value = 1e-10). Meanwhile, SLEEPER gene family members were identified by hmmer3 software (http://hmmer.org/) (e-value = 1e-5), based on HMM of the conserved domains zf-BED (PF02892) and Dimer_Tnp_hAT (PF05699) from Pfam Database (http://pfam.xfam.org/). We later merged the blast alignment result and the HMM search result. The duplicate values were removed.
Phylogenetic tree construction
To understand the evolutionary relationships among SLEEPER genes family in B. napus, 162 full-length SLEEPER protein sequences were extracted to generate a phylogenetic tree by MEGA-X, with DAYSLEEPER and CYTOSLEEPER protein in (A) thaliana, 93 SLEEPERs in (B) oleracea, 51 in B. rapa and 34 in O. sativa (Table S1) selected as representatives. Multiple sequence alignments were subjected to a maximum likelihood (ML) method for the construction of the phylogenetic tree. The tree was then visualized by Evolview (http://www.evolgenius.info/evolview/). According to the evolutionary relationships, the nomenclature of B. napus SLEEPERs were carried out using standardized gene nomenclature for the cruciferous species. Similarities and identities among all the SLEEPERs proteins in the ML tree were analyzed by SIAS tools (http://imed.med.ucm.es/Tools/sias.html).
Gene structures, motifs, conserved domains and promoter region cis-elements analysis
The conserved motif structures were analyzed by MEME online tool (http://meme-suite.oorg/index.html). We obtained the conserved domain and analyzed the motif function through NCBI-CDD database. The 1500 bp sequences of ATG upstream of SLEEPERs were extracted as the promoters. PlantCare (http://bioinformatics.psb.ugent.be/webtools/pantcare/html/) was used to analyze the cis-elements of promoters.
All the data above was visualized by TBtools [36].
Chromosomal location, distribution and collinearity, Ka/Ks analysis
The positions of SLEEPERs on chromosomes were displayed by TBtools. The Multiple Collinearity Scan (MCScanX) was used to analyze the syntenies and collinearity of SLEEPER Genes in B. napus, B. oleracea and B. rapa [37]. The version of Genomic sequence and GFF Annotated feature files were BOL and CAAS_Brap_v3.01 from NCBI. Ka, Ks calculations were performed with the “Ka/Ks Calculator” from TBtools, and the Circos plots were constructed by “Advanced Circos” in TBtools [38].
Expression analysis of BnSLEEPERs
We used RNA-Seq datasets previously published of line 3529 by our lab to analyze the expression patterns of SLEEPERs in various tissues of B. napus at different stages, including root, stem and leaf tissues at the seedling stage, root, stem, flower and leaf tissues at the flowering stage, and silique and leaf tissues at the silique stage. For each tissue sample, there were two biological replicates, i.e. two technical replicates were set up for each sequencing sample of B. napus in the same growth state and the same environment.
Clean reads from raw data were compared with the reference genome of ZS11. To facilitate the analysis of differences between all samples, Transcripts Per Million (TPM) was used as the standardized expression value. The TPM of SLEEPER genes were retrieved and then used to draw the heatmap after log transformation. DESeq2 software was used to analyze the differential expression of genes based on reads count. The screening criteria for differentially expressed genes (DEGs) were: False Discovery Rate (FDR) < 0.05 and |log2FoldChange| ≥ 3. FoldChange represents the ratio of FPKM between two samples, the raw data of transcriptome sequencing was upload in NCBI database (PRJNA932529, https://dataview.ncbi.nlm.nih.gov/object/PRJNA932529?reviewer=gqeda6eelsvr5bg9g06f911c19).
We next used quantitative real-time PCR (qRT-PCR) analysis to test the data of RNA-seq. The qRT-PCR was performed on two biological replicates and three technical replicates to analyze expression profile of BnDAYSLEEPER8, BnRAPE2.12, 3.38 and 4.35 using qPCR SYBR Green Master Mix Kit (YEASEN Biotech). The procedure was conducted as follows: qPCR cycle parameters were set as 95 °C for 30s, 39 cycles of 5s at 95 °C, 15s at 57 °C, a final melting curve from 65 to 95 °C in 0.5 °C increments. The relative gene expression levels were calculated using the 2−ΔΔCt method with β-actin from B. napus.
The comparison of RNA-seq and qRT-PCR data were performed by Student’s t-test and Pearson correlation coefficient (Table S10).
Prediction of isoelectric point, signal peptide, transmembrane helix and subcellular localization of BnSLEEPERs
Isoelectric point (PI) was predicted by Protparam (http://web.expasy.org/protparam/), signal peptide was predicted by SignalP 5.0 Sever (https://services.healthtech.dtu.dk/service.php?SignalP-5.0). Transmembrane helix was predicted by TMHMM Sever v.2.0 (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0). Subcellular localization was predicted by Plant-mPLoc (http://www.csbio.situ.edu.cn/bioinf/plant-multi/).
In this study, vector pBI221-eGFP was used for subcellular localization of BnDAYSLEEPER8, BnRAPE2.12, 3.38 and 4.35, these four genes were ligated to vector pBI221 before eGFP reporter gene and driven by CaMV35S promoter. Restriction sites and primers used in this part are listed in Supplementary Table S8.
Then, 4-week-old N. benthamiana leaves in good growth condition were cut into slim strips and then incubated in the enzymatic solution for 3 h at 30 °C in the dark. One-fold volume of pre-cooled solution containing W5 was added after removing the residue in the protoplast preparation solution with forceps. After being filtered and centrifuged, the same volume of pre-cooled W5 solution was added again for 1 h in an ice bath. The protoplasts were collected in the precipitate obtained by centrifugation of the solution. 30ug of the recombinant plasmid, 200µL of protoplasts solution and 230µL of 40% PEG solution were gently mixed in a 2mL tube and left at room temperature for 15 ~ 20 min before the transformation was terminated by adding 4 folds volume of W5 solution. After centrifuging the solution at 100rmp for 2 min, the precipitate containing the protoplasts was washed in 1 mL W5 solution, and then resuspended with 1 mL WI. The transformed protoplasts were incubated in the dark at 25℃ for 6 ~ 8 h to express the fusion protein. Finally, we used a confocal laser microscope (Leica microsystems DM4 B, the excitation wavelength is 488 nm) to observe the protoplasts. The composition of all solutions is in the Table S7.
Data availability
The datasets generated and/or analyzed during the current study are available in the Sequence Read Archive (SRA) repository with the BioProject accession, PRJNA932529. (https://dataview.ncbi.nlm.nih.gov/object/PRJNA932529?reviewer=gqeda6eelsvr5bg9g06f911c19)
Abbreviations
- CREs:
-
Cis-acting elements
- DEG:
-
Differentially expressed genes
- FDR:
-
False discovery rate
- FPKM:
-
Fragments per kilobase of exon model per million reads mapped
- hAT:
-
Hobo, Activator, and Tam3
- NLS:
-
Nuclear localization signal
- ORFs:
-
Open reading frames
- PI:
-
Isoelectric point
- RAPE:
-
RAPESLEEPER
- SP:
-
Signal peptide
- TEs:
-
Transposable elements
- TH:
-
Transmembrane helices
- TIR:
-
Terminal inverted repeat
- TPM:
-
Transcripts per million
- ZBED:
-
BED-zinc finger transcription factors
- zf-BED:
-
BED-zinc finger domain
References
Doolittle WF, Sapienza C. Selfish genes, the phenotype paradigm and genome evolution. Nature. 1980;284:601–3. https://doi.org/10.1038/284601a0.
The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. https://doi.org/10.1038/35048692.
Shu O, et al. The map-based sequence of the rice genome. Nature. 2005;436:793–800. https://doi.org/10.1038/nature03895.
Schnable PS, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–5. https://doi.org/10.1126/science.1178534.
Jurka J, et al. Repetitive sequences in complex genomes: structure and evolution. Annu Rev Genomics Hum Genet. 2007;8:241–59. https://doi.org/10.1146/annurev.genom.8.080706092-416.
David, et al. Ty element transposition: reverse transcriptase and virus-like particles. Cell. 1985;42:507–17. https://doi.org/10.1016/0092-8674(85)90108-4.
Ochmann MT, Ivics Z. Jumping ahead with sleeping beauty: mechanistic insights into cut-and-paste transposition. Viruses. 2021;13:76. https://doi.org/10.3390/v13010076.
Kapitonov VV, Jerzy J. Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet. 2007;23:521–9. https://doi.org/10.1016/j.tig.2007.08.004.
Peng Y, et al. Elimination of a retrotransposon for quenching genome instability in modern rice. Mol Plant. 2019;12:1395–407. https://doi.org/10.1016/j.molp.2019.06.004.
Li Y, et al. Domestication of transposable elements into microrna genes in plants. PLoS ONE. 2011;6:e19212. https://doi.org/10.1371/journal.pone.0019212.
Kojima KK, Jerzy J. Crypton transposons: identification of new diverse families and ancient domestication events. Mob DNA. 2011;2:12. https://doi.org/10.1186/1759-8753-2-12.
Hudson ME et al. 2003, The FHY3 and FAR1 genes encode transposase-related proteins involved in regulation of gene expression by the phytochrome A-signaling pathway, Plant J., 34, 453–471. https://doi.org/10.1046/j.1365-313x.2003.01741.x
Carmona LM et al. 2016, Collaboration of RAG2 with RAG1-like proteins during the evolution of V(D)J recombination, genes dev., 30, 909–17. https://doi.org/10.1101/gad.278432.116
Rodriguez-Moreno A. Characterization of an eutherian gene cluster generated after transposon domestication identifies Bex3 as relevant for advanced neurological functions. Genome Biol. 2020;21:267–94. https://doi.org/10.1186/s13059-020-02172-3.
Mao D, et al. The harbinger transposon-derived gene PANDA epigenetically coordinates panicle number and grain size in rice. Plant Biotechnol J. 2022;20:1154–66. https://doi.org/10.1111/pbi.13799.
Rubin E, et al. Structure and evolution of the hAT transposon superfamily. Genetics. 2001;158:949–57. https://doi.org/10.1093/genetics/158.3.949.
Kempken F, Windhofer F. The hAT family: a versatile transposon group common to plants, fungi, animals, and man. Chromosoma. 2001;110:1–9. https://doi.org/10.1007/s004120000118.
Knip M, et al. The SLEEPER genes a transposase-derived angiosperm-specific gene family. BMC Plant Biol. 2012;12:192–207. https://doi.org/10.1186/1471-2229-12-192.
Rizkallah R, et al. Global mitotic phosphorylation of C2H2 zinc finger protein linker peptides. Cell Cycle. 2011;10:3327–36. https://doi.org/10.4161/cc.10.19.17619.
Sun T, et al. Two hAT transposon genes were transferred from Brassicaceae to broomrapes and are actively expressed in some recipients. Sci Rep. 2016;6:30192. https://doi.org/10.1038/srep30192.
Yellan I, et al. Diverse eukaryotic CGG-binding proteins produced by independent domestications of hAT transposons. Mol Biol Evol. 2021;38:2070–5. https://doi.org/10.1093/molbev/msab007.
Hussain A, et al. A genome-wide comparative evolutionary analysis of zinc finger-BED transcription factor genes in land plants. Sci Rep. 2022;12:12328. https://doi.org/10.1038/s41598-022-16602-8.
Bundock P, Hooykaas P. An Arabidopsis hAT-like transposase is essential for plant development. Nature. 2005;436:282–4. https://doi.org/10.1038/nature03667.
Knip M, et al. DAYSLEEPER: a nuclear and vesicular-localized protein that is expressed in proliferating tissues. BMC Plant Biol. 2013;12:211–24. https://doi.org/10.1186/1471-2229-13-211.
Chen X, et al. A high-quality Brassica napus genome reveals expansion of transposable elements, subgenome evolution and disease resistance. Plant Biotechnol J. 2021;19:615–30. https://doi.org/10.1111/pbi.13493.
Hurst LD. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 2002;18:486–95. https://doi.org/10.1016/s0168-9525(02)02722-1.
Chalhoub B, et al. Erratum: early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science. 2014;345:950–3. https://doi.org/10.1126/science.1253435.
Liu S, et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun. 2014;23:3930–4. https://doi.org/10.1038/ncomms4930.
Cannon SB, et al. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004;1:10–3. https://doi.org/10.1186/1471-2229-4-10.
Xie M, et al. Genome-wide identification and comparative analysis of CLE family in rapeseed and its diploid progenitors. Front Plant Sci. 2022;13:998082. https://doi.org/10.3389/fpls.2022.998082.
Cheng F, et al. Gene retention, fractionation and subgenome differences in polyploid plants. Nat Plants. 2018;4:258–68. https://doi.org/10.1038/s41477-018-0136-7.
Markljung E, et al. ZBED6, a novel transcription factor derived from a domesticated DNA transposon regulates IGF2 expression and muscle growth. PLoS Biol. 2009;7:e1000256. https://doi.org/10.1371/journal.pbio.1000256.
Saghizadeh M, et al. ZBED4, a BED-type zinc-finger protein in the cones of the human retina. Invest Ophthalmol Vis Sci. 2009;50:3580–8. https://doi.org/10.1167/iovs.08-2751.
Hayward A, et al. ZBED evolution: repeated utilization of DNA transposons as regulators of diverse host functions. PLoS ONE. 2013;8:e59940. https://doi.org/10.1371/journal.pone.0059940.
Somerville TDD, et al. ZBED2 is an antagonist of interferon regulatory factor 1 and modifies cell identity in pancreatic cancer. Proc Natl Acad Sci. 2020;117:11471–82. https://doi.org/10.1073/pnas.1921484117.
Chen CJ, et al. An integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13:1194–202. https://doi.org/10.1016/j.molp.2020.06.009.
Wang YP, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. https://doi.org/10.1093/nar/gkr1293.
Chen CJ et al. 2022, A painless way to customize circos plot: from data preparation to visualization using tbtools, iMeta, 1, e35. https://doi.org/10.1002/imt2.35
Acknowledgements
We would like to acknowledge all researchers in our laboratory for their help.
Funding
The National Key Research and Development Program of China (2023YFD1200202), the Provincial Science and Technology Department project (2022ZDZX005 and 2021YFYZ0018-LH) and the Deyang municipal Key Research and Development Project (2021NZ043).
Author information
Authors and Affiliations
Contributions
R.Z., S.A., Yun Zhao and M.W. conceived and designed the experiments, R.Z., S.A., J.F., S.L., Y.F. and Ying Zhang performed the experiments. R.Z., S.A., J.F. and R.W analyzed the data. R.Z., M.W contributed reagents/materials/analysis tools. R.Z., S.A. and M.W wrote the main manuscript text. All authors have reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
Our research did not involve any human or animal subjects, materials, or data. We owned the plant materials and we have permissions to use them. We declare that the collection and use of plant materials in this study complies with relevant institutional, national, and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, R., An, S., Fu, J. et al. Genome-wide identification and characterization of SLEEPER, a transposon-derived gene family and their expression pattern in Brassica napus L.. BMC Plant Biol 24, 810 (2024). https://doi.org/10.1186/s12870-024-05544-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-05544-0