
Abstract
Background
Punica granatum is a fundamentally important fruit tree that has important economic, medicinal and ornamental properties. At present, there are few reports on the mitochondrial genome of pomegranate. Hence, in this study the P. granatum mitogenome was sequenced and assembled to further understanding of organization, variation, and evolution of mitogenomes of this tree species.
Results
The genome structure was multi-chromosomes with seven circular contigs, measuring 382,774 bp in length with a 45.91% GC content. It contained 74 genes, including 46 protein-coding genes, 25 tRNA genes, and three rRNA genes. There were 188 pairs of dispersed repeats with lengths of 30 or greater, primarily consisting of reverse complementary repeats. The mitogenome analysis identified 114SSRs and 466 RNA editing sites. Analyses of codon usage, nucleotide diversity and gene migration from chloroplast to mitochondrial were also conducted. The collinear and comparative analysis of mitochondrial structures between P. granatum and its proximal species indicated that P. granatum ‘Taishanhong’ was closely related to P. granatum ‘Qingpitian’ and Lagerstroemia indica. Phylogenetic examination based on the mitogenome also confirmed the evolutionary relationship.
Conclusion
The results offered crucial information on the evolutionary biology of pomegranate and highlighted ways to promote the utilization of the species’ germplasm.
Similar content being viewed by others


Introduction
Pomegranate (Punica granatum L.) belongs to the genus Punica of the Lythraceae family, and is an essentially valuable fruit tree in the world [1,2,3]. It is native to Central Asia region and grows mainly in tropical and subtropical countries such as India, Iran, China, and the USA [4, 5]. The consumption of pomegranates has shown a remarkable increase due to their exceptional economic, nutritional, medicinal and ornamental value. The different parts (fruit peel, juice, seeds, flowers, leaves, bark) are rich in bioactive constituents, and have various medicinal advantages to provide high antioxidant, antineoplastic, anticancer and phytonutrient capacity [6,7,8,9]. At present, pomegranate related industries are expanding rapidly, and plays an important role in the economic, cultural and ecological fields.
Mitochondria are semi-autonomous organelles with a mitochondrial genome found in virtually all eukaryotic cells, which synthesize Adenosine Triphosphate (ATP) through the tricarboxylic acid cycle and oxidative phosphorylation to provide energy for cells [10, 11]. Mitochondria are involved in numerous metabolic processes and perform crucial roles in cell differentiation, apoptosis, cell development, and cell division by converting biomass energy into chemical energy for daily activities [12,13,14]. Plant chloroplast genomes have revealed conserved genome structure, but the mitochondrial genome has very low collinearity due to extensive rearrangement and repeat sequences, whereas its protein-coding genes are relatively conserved [12, 15]. Plant mitogenomes are species specific and possess many unique features, which vary considerably in length, gene order, and gene content [16, 17]. The structures of mitogenomes are normally depicted as a complex and dynamic circular structure with a large amount of unconserved DNA of unknown function [18]. In some species, the mitochondrial genomes have linear conformations, branched and multichromosomal structures [14, 19]. The above characteristics of the mitochondrial genome provide helpful information for evolutionary and phylogenetic studies.
The mitochondrial genomes of 545 plant species have been assembled and reported [14], including Sapindaceae [17], Rosaceae [20], Anaeardiaceae [21], and Myrtaceae [22] family. Mitochondrial genomes of some fruit crops have been reported, such as malus domestica [23], Satsuma mandarin [24], and kiwifruit [25], etc. While the chloroplast genomes of approximately 114 species of P. granatum are available in the NCBI database (as of 28 June 2023), only one pomegranate variety’s mitochondrial genome has been identified and published [26]. The germplasm resources of pomegranate are abundant, but the mitochondrial genome differences between different varieties are still unclear. Therefore, more in-depth researches of the mitochondrial genome of P. granatum are of great significance for its effective utilization and genetic improvement.
In this study, we sequenced, assembled and annotated the complete mitochondrial genome of main P. granatum cultivar ‘Taishanhong’. The mitogenome characteristics, repetitive sequences, SSR identification, and RNA editing prediction were investigated. Further analyses regarding species synteny and phylogeny were carried out for the determination of phylogenic positions and molecular diversity of the myrtales. The gene transformation of chloroplast genome to mitochondrion DNA was analyzed. This study is anticipated to lay the foundation for a deeper understanding and exploration of evolutionary variations and genomic breeding of pomegranate mitochondrial genomes.
Results
Genomic features of P. granatum mitochondrial genome
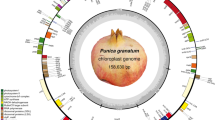
The P. granatum mitochondrial genome was sequenced, assembled, and annotated, which has been archived in GenBank under the accession numbers OQ973289, OQ973290, OQ973291, OQ973292, OQ973293, OQ973294 and OQ973295. The mitochondrial genome of P. granatum was assembled into 7 circular contigs with lengths ranging from 3,728 bp to 126,124 bp, with a total length of 382,774 bp (Fig. 1, Fig. S1 and Fig. S2). The total GC content was 45.91%, ranging between 45.42% and 52.60% (Table S1). It encoded 74 genes, including 46 mitochondrial protein-coding genes, 25 tRNA genes, and three rRNA genes (rrn5, rrn18, and rrn26). The total GC contents of CDS, tRNA and rRNA genes were 42.35%, 50.85% and 52.14%, respectively (Table S1).

Circular Map of the P. granatum mitochondrial genome. The genome consisted of 7 circular chromosomes. The genes that code in the forward direction are on the outside of the circle, and the genes that code in the opposite direction are on the inside of the circle. The inside gray circle represents GC content
The protein-coding genes included 34 core genes and nine variable genes (Table 1). The core genes included five ATP synthase genes (atp1, atp4, atp6, atp8 and atp9), nine NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7 and nad9), four cytochrome c biogenesis genes (ccmB, ccmC, ccmFC and ccmFN), three cytochrome c oxidase genes (cox1, cox2 and cox3), one Maturases gene (matR), one membrane transport protein gene (mttB) and one ubichinol cytochrome c reductase gene (cob). The variable genes included four large subunits of ribosomal protein (rpl10, rpl16, rpl2 and rpl5), seven small subunits of ribosomal protein (rps1, rps3, rps4, rps7, rps12, rps14 and rps19), and one succinate dehydrogenase (sdh4). Among these, the genes atp9, mttB, nad2 and nad5 had two copies. The nad1, nad2, nad5 and nad7 genes contained four introns and nad4 gene had three introns, while ccmFC, cox2, rpl2 and rps3 had one intron. The trnC-GCA, trnM-CAT, trnP-TGG and trnS-TGA genes were multi-copy genes. The trnA-TGC, trnI-GAT and trnI-TAT genes contained one intron.
Condon usage and RSCU analysis
The ATG was the most frequent start codon for protein-coding genes, while the nad1 gene was an exception, having the initiating codon ACG (ATG). The stop codons TAA, TAG, TGA, and CGA (TGA) were identified (Table 1). These results indicated that the C to U RNA editing phenomenon was found in the start or stop codons.
The relative synonymous codon usage (RSCU) analysis of P. granatum mitochondrial genome was shown in Fig. 2. It contained 10, 445 codons excluding termination codons in protein-coding genes regions. The most frequent codons used were UUU (Phe), AUU (Ile) and GAA (Glu) and were used>300 times, while CUG (Met), UUG (Met) and UAG (Ter) were rarely found (Table S2). We found that most RSCU values of the codons ending with A or T were higher than 1.0, while most of those ending with C or G had RSCU values of less than 1. Codon usage was generally strongly biased toward A or T (U) at the third codon position in the P. granatum mitogenome. Similar results had been found in the mitogenome of other plants [17, 27].

Relative synonymous codon usage (RSCU) in the P. granatum mitochondrial genome. The different amino acids are shown on the x-axis. RSCU values are the number of times a particular codon is observed relative to the number of times that codon would be expected for a uniform synonymous codon usage. The bottom square represents all the codons that code for each amino acid, and the height of the top column represents the sum of all the codon RSCU values
Repeat sequence analysis
Dispersed repeats are repeats that are distributed at different locations in the genome. A total of 188 pairs of repeats with a length greater than or equal to 30 bp were found, including 97 pairs of reverse complementary repeats, 91 pairs of forward repeats. The longest reverse complementary repeat was 355 bp, while the longest forward repeat was 24,984 bp (Table S3).
The simple repeated sequences (SSRs) with motifs of one to six bases are abundant in higher plants. In the study, a total of 141SSRs were detected in the P. granatum mitogenome, including 58 (41.13%) mono-, 19 (13.48%) di-, 18 (12.77%) tri-, 35(24.82%) tetra-, 9 (6.38%) penta-, and 2 (1.42%) hexanucleotide repeats (Fig. 3A). Among them, monomeric and dimeric SSRs accounted for 54.61% of the total SSRs. Mononucleotide repeats of A/T (34.75%) were more prevalent than the other repeat types, and dinucleotide repeats of AG/CT (7.80%) were the second most numerous.

The number of SSR motifs and RNA editing sites predicted in P. granatum mitogenome. (A) The number of identified SSR motifs. (B) The number of RNA editing sites identified in each PCGs
Prediction of RNA editing sites
RNA editing events were identified for 36 unique PCGs based on online website predictions. There were 466 potential RNA editing sites distributed among all PCGs, and all of which were C-to-U base editing (Fig. 3B and Table S4). The nad4 gene was the most edited and possessed 39 potential RNA editing sites of the mitochondrial genes. This was followed by the ccmB gene with 36 RNA editing events. The edited number of rps1 gene was the lowest and had only one potential RNA editing events among all mitochondrial genes.
The total number of hydrophilic-hydrophobic type induced by RNA editing was 224 sites, which had the highest proportion at 48.07% (Table 2). The hydrophobic-hydrophobic and hydrophilic-hydrophilic type was 31.33% (146 sites) and 12.23% (57 sites), respectively. The hydrophilic-stop number was the lowest and the proportion was 1.07% (5 sites). Among them, there were 10 site conversions of CCT to TTT and 8 conversions of CCC to TTC.
Nucleotide diversity and comparative analysis of mitochondrial structure
The nucleotide diversity (pi) values of 38 regions were calculated and ranged from 0 to 0.10582, with an average of 0.03132 (Fig. 4A and Table S5). The pi value of gene10.nad9 region was highest in these regions, which was 0.07259 and 0.0698 in gene37.atp9 and gene33.rrn18 regions, respectively. The lower pi values suggested that the mitogenome sequences of P. granatum were highly conserved.

Nucleotide diversity and comparative analysis of P. granatum mitochondrial structure. (A) The gene nucleotide variability (pi) values. (B) Comparative analysis of mitochondrial structure of P. granatum and proximal species. The outer two circles describe the length and direction of the genome. The circles inside represent similar results compared to other reference genomes. The black circles represent the GC content
The mitogenome structures of P. granatum and its proximal species belonging to the Myrtales were comparatively analyzed using CGVIEW software (Fig. 4B). There were high similarities in the mitochondrial structure of P. granatum and Eucalyptus grandis (NCBI Number: MG925370.1), Lagerstroemia indica (NCBI Number: KX641464.1) and Medinilla magnifica (NCBI Number: MT043351.1).
Phylogenetic and collinearity analysis
The mitochondrial genome of proximate species of pomegranate is rarely reported. To further explore the evolutionary relationships of P. granatum, the phylogenetic tree was constructed by maximum likelihood method (Fig. 5). Phylogenetic analysis was performed on 30 conserved mitochondrial PCGs (atp1, atp4, atp6, atp8, atp9, ccmC, ccmFc, ccmFn, cob, cox1, cox2, cox3, matR, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9, rpl5, rps1, rps3, rps4, rps12, rps14 and sdh4) from11 species. The mitochondrial genomes of Malus domestica, Vitis vinifera and Fragaria orientalis were set as outgroups. The phylogenetic tree strongly supports (100% bootstrap support) the close phylogenetic relationship between P. granatum ‘Taishanhong’ and P. granatum ‘Qingpitian’ and Lagerstroemia indica. The result indicated that the evolutionary relationship of mitochondrial genome among different pomegranate varieties is relatively close.

The evolutionary relationship between P. granatum and homologous species
The collinearity analysis of Lagerstroemia indica, Eucalyptus grandis, Medinilla magnifica and P. granatum were performed with two methods (Fig. 6). The mitogenome of P. granatum and Lagerstroemia indica had more collinear forward alignment and reverse complementary alignment sequences, which was followed by P. granatum and Eucalyptus grandis (Fig. 6A). There were many homologous collinear blocks between the P. granatum mitochondrial genome and other three proximate species (Fig. 6B). Some gaps illustrate that these sequences are unique to the species and have no homology with the rest of the species. The results suggest that the P. granatum mitochondrial genome has undergone a lot of genomic rearrangement with close species.

Collinear analysis of mitochondrial sequence with two methods. (A) The horizontal coordinate in each box indicates the assembled sequence, and the vertical coordinate indicates the other sequences. The red line in the box indicates the forward alignment, and the blue line indicates the reverse complementary alignment. (B) The boxes in each row represent a genome, and the lines in the middle represent homologous regions
Intracellular gene transfer of P. granatum organelle genomes
The P. granatum mitogenome sequence was approximately 2.41 times longer than its cp. genome (NCBI Number: MK603511, 1158,638 bp). According to sequence similarity analysis, there were 22 homologous fragments between the mitogenome and chloroplast genome, with a total length of 19,889 bp, accounting for 5.20% of the total mitogenome (Fig. 7 and Table S6). By annotating these homologous sequences, 11 complete genes were also found on 22 homologous fragments, including nine tRNA genes (trnD-GUC, trnH-GUG, trnI-GAU, trnM-CAU, trnP-UGG, trnS-UGA, trnV-GAC, trnW-CCA, trnN-GUU) and two rRNA genes (rrn5 and rrn16) (Table S6). There were 35 gene sequences transferred from the chloroplast to the mitochondria in P. granatum (Table S7). The length of atpA_len507 gene was the longest with a length of 471 bp. The ndhA_len363 gene had the shortest length (35 bp).

Homologous fragments of chloroplast and mitochondrial sequence in P. granatum. Chloroplast is chloroplast sequence and chr is mitochondrial sequence. Genes from the same complex are labeled with the same color, with homologous sequences represented at the center line junctions
Discussion
Structure and characteristic of P. granatum mitogenome
The genome structure of plant mitochondrial genomes has always been mapped as a single master circle and a collection of subgenomic circles because of the large, direct, repeat recombination [28, 29]. For example, the mitochondrial genomes of bitter gourd [11] and Vitex rotundifolia [30] were mapped as a molecular circle. The mitochondrial genome of Chaenomeles speciosa was multi-chromosomes with two circular contigs [31] and Paphiopedilum micranthum with 26 circular contigs [32], while the Q. acutissima mitogenome was a branched structure of two circular molecules and one linear molecule [33]. In the present study, the P. granatum ‘Taishanhong’ mitogenome was represented by seven circular contigs structure (Fig. 1) with a total length of 382,774 bp. There was significant difference in the structure (a single linear molecule) and size (404,807 bp) of P. granatum ‘Qingpi Sweet’ mitogenome [26]. The difference in genome size may be related to the different mitochondrial structure of the two varieties. This suggested that the mitochondrial genomes of pomegranate were species specific and possessed many unique features, which is consistent with research on other species [17, 21]. The result provided new insights into the function and structure of the mitochondrial genomes in pomegranate species.
GC content is an important factor for assessing species. The total GC content of P. granatum mitogenome was 45.91%, which was highest in the rRNA genes. The result was comparable to that of Phaseolus vulgaris (45.11%) [16], Bitter Gourd (45.60%) [11], and Bupleurum chinense (45.68) [34], which was higher than that in Mangifera sylvatica (44.66%) [21]. The mitogenome of P. granatum ‘Taishanhong’ encoded 74 genes, including 46 protein-coding genes, 25 tRNA genes, and three rRNA genes, which was higher than that in P. granatum ‘Qingpi Sweet’(60 genes) [26].
The intron contents of the plant mitochondrial genes are conserved in each lineage but are variable among different lineages [35]. In the present study, the trans-spliced genes nad1, nad2, nad5, nad7 contained four introns, and nad4 gene had three introns. These trans-spliced introns were postulated to have evolved from one common ancestor by fragmentation of a cis-spliced arrangement [36].
Identification of condon usage and repeat sequences
Most PCGs were the typical ATG start codon, while nad1 gene uses the ACG as initiation codons in the P. granatum mitogenome. The phenomenon also has been found in other studies, which were considered to be altered by RNA editing modification [37, 38].
Codon usage preference is usually considered to be the result of the tendency to develop relative equilibrium within cells during species evolution, and codons with a RSCU greater than 1 are considered to be used by amino acid preference [17, 39,40,41]. The result indicated that Phe, Ile and Glu were the most common amino acids, and Met and Ter were much less common in the P. granatum mitogenome. The most prevalent amino acids were Ala, Tyr, and His in Camellia sinensis [42], while Ala, Gln, and His were the most common amino acids in Clematis acerifolia [43].
Repeated sequences are widely present in the mitochondrial genome, which are generally vital for intermolecular recombination, structural variations and extreme mitogenome sizes [44,45,46]. In this study, a total of 188 pairs of repeats were observed, including 97 pairs of reverse complementary repeats and 91pairs of forward repeats. These repetitive sequences play a crucial role in the assembly of the P. granatum mitogenome [47]. Simple sequence repeats are a special kind of presence among tandem repeats, which can better reflect species’ genetic structure and genetic diversity changes [48, 49]. There was a total of 141 SSRs were detected and mononucleotide repeats of A/T were the most frequent, which was consistent with other research findings in Momordica charantia [11] and Calla Lilies [28]. It provided a large number of reference markers for the evaluation of pomegranate germplasm diversity and species identification.
RNA editing analysis
RNA editing is a very common phenomenon in the mitochondrial genomes of higher plants. Almost all of the transcription products of mitochondrial PCGs are subjected to varying degrees of RNA editing but rarely occur in rRNA, tRNA, and introns [50,51,52]. RNA editing can affect the composition of the final mitochondrial proteome, which can create start and stop codons in the mitochondrial mRNA molecules [53, 54]. Previous researches had uncovered approximately 421RNA editing sites within 26 genes in Acer truncatum [17] and 486 RNA editing sites within 31 genes in Phaseolus vulgaris [16]. There were 466 potential RNA editing sites distributed among 36 PCGs, and all were C-to-U base editing in P. granatum mitogenome. The result was similar to the study in Salvia officinalis [55], Leucaena trichandra [56] and Pereskia aculeata [57]. The nad4 gene possessed 39 potential RNA editing sites and was the most edited of pomegranate mitogenome, which was supported by other studies [14, 41].
Phylogenetic relationships and synteny analysis
Changes in the size and structure of plant mitochondrial genomes are evident, but functional genes remain conserved [58]. The nucleotide diversity (pi) reveals the variation of nucleotide sequence in different species (Fig. 4A). Our data showed that the pi value of nad9 gene region was highest in these regions. The lower pi values suggested that the mitogenome sequences of P. granatum were highly conserved.
There were high similarities in the mitochondrial structure of P. granatum and Eucalyptus grandis, Lagerstroemia indica and Medinilla magnifica (Fig. 4B). The phylogenetic tree strongly supports the close phylogenetic relationship between P. granatum and Lagerstroemia indica. It was further confirmed from the molecular level that the pomegranate belonged to the Lythraceae family [59]. The co-linearity between P. granatum and Lagerstroemia indica was also high with large homozygous co-linear blocks, suggesting that the two species have undergone extensive rearrangement phenomena in the long evolutionary process [60]. The result provided an effective way to understand species evolution of pomegranate, and the specific mechanism and process need to be further studied.
Intracellular gene transfer of P. granatum organelle genomes
Gene transfer from chloroplasts to mitochondrial genomes is common during long-term plant evolution. The discovery of intracellular gene transfers can contribute to the accurate assembly of chloroplast and mitochondrial genomes and to the understanding of organelle genome evolution [61, 62]. We found 22 homologous fragments between the pomegranate mitogenome and chloroplast genome, with a total length of 19,889 bp, accounting for 5.20% of the total mitogenome. There were 35 gene sequence of chloroplast transfer to mitochondria in P. granatum, while the length of atpA_len507 gene was the longest with a length of 471 bp. Previous studies have found that tRNA genes occasionally migrated from the chloroplast to mitochondria, such as trnD-GUC, trnN-GUU, and trnH-GUG in Acer truncatum [17], trnV-GAC, trnP-TGG, trnI-GAU, and trnA-UGC in Camellia sinensis [42], etc. The nine complete tRNA genes of homologous fragments were identified from the chloroplast of P. granatum, which suggested that they may play a role in normal functions [63]. Therefore, larger-scale sampling is required better to understand the evolution of phylogenetic analyses and molecular markers of pomegranate. These studies will provide a substantial foundation for understanding pomegranate mitogenome evolution and facilitate mitochondrial-based breeding.
Conclusions
In this study, we assembled and annotated the complete mitogenome of P. granatum cultivar ‘Taishanhong’. The genome structure was multi-chromosomes with seven circular chromosomes, and total length was 382,774 bp and GC content was 45.91%. In addition, the codon usage, sequence repeats, RNA editing and chloroplast to mitochondrion DNA transformation were also analyzed. There were high similarities in the mitochondrial structure between P. granatum and its proximal species. Evolutionary and collinear analysis showed that P. granatum was closely related to P. granatum ‘Qingpitian’ and Lagerstroemia indica. This study provided information on the genetic characteristics, phylogenetic relationships, species identification and biological evolution of P. granatum.
Methods and materials
Plant materials
The tissue culture seedlings of P. granatum cultivar ‘Taishanhong’ were planted in the tissue culture room at Shandong Institute of Pomology (Taian, Shandong, China). Plants were kept in the dark for 14 d to obtain etiolated seedlings. Then, the fresh leaves were obtained and cleaned with DEPC water and kept in a freezer at -80 °C.
DNA extraction and sequencing
The second and third generation sequencing strategy was adopted to complete the assembly of mitochondrial genome [55]. The total genomic DNA was extracted by a DNA plant extraction kit (Tiangen, Beijing, China). After the genomic DNA of the sample was qualified, the DNA was fragmented using ultrasound. DNA fragments were purified, terminal repair, A was added to the 3 ‘end, and sequencing splices were connected. The fragment size was selected by agarose gel electrophoresis and PCR amplification was performed to form sequencing library. The constructed libraries were inspected first, and second qualified libraries were sequenced using Illumina Novaseq6000 platform by Nanjing Genepioneer Biotechnologies (Nanjing, China). The sequencing was performed as paired-end and read length was 150 bp. The Fastp (version 0.20.0, https://github.com/OpenGene/fastp) was used to filter the raw data, removing primer sequences and sequencing junctions, discarding reads with quality scores below Q5, and removing N values greater than 5. Finally, we obtained a total of 21, 083,687clean reads and the GC content was 40.21%. The Q20 and Q30 were respectively 96.39% and 90.68%.
After the samples passed the quality inspection, the Third generation sequencing experiment was performed. The genomic DNA is randomly interrupted, and large fragments of DNA are enriched and purified by magnetic beads. Then, the large fragments are cut and recovered, and the damage of fragmented DNA is repaired. After purification, the end repair was performed on both ends of the DNA fragment and A was added. The joint in the SQK-LSK109 connection kit was used for the connection reaction. Finally, the constructed DNA library was quantitatively detected. After the DNA library with a certain concentration and volume was added into the Flow cell, and was transferred to the Oxford Nanopore PromethION sequencer for real-time single molecule sequencing. The three generations of sequencing data were filtered using filtlong v0.2.1 software, and counted using Perl scripts.
Assembly and annotation of the mitochondrial genome
The minimap2 v2.1 software was used to compare the reference gene sequence (plant mitochondrial core gene, the company’s own core genetics website: https://github.com/xul962464/plant_mt_ref_gene) with the original third-generation data [64]. The sequences with alignment length greater than 50 bp were selected as candidate sequences, and the sequences with more alignment genes and higher alignment quality were selected as seed sequences. The sequences with overlap greater than 1 kb and similarity greater than 70% were screened and obtained all third-generation sequencing data of mitochondrial genome. The canu software [4] was used to correct the obtained third-generation data and bowtie2 (v2.3.5.1) was used to compare the second-generation data to the corrected sequence [65]. The Unicycler (v0.4.8) default parameter comparison was used to concatenate the second-generation data and the third-generation data after correction. The corrected third-generation sequencing data was compared to the contig obtained in the second step of Unicycler using minimap2, and the branch direction was manually determined to obtain the final assembly result.
The encoded proteins and rRNAs were compared to published plant mitochondrial sequences using BLAST, and further manual adjustments were made based on closely related species. The tRNA was annotated using tRNAscanSE (http://lowelab.ucsc.edu/tRNAscan-SE/) software [66]. ORFs were annotated using Open Reading Frame Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). The minimum length was set as 102 bp, and redundant sequences and sequences with overlap with known genes were excluded. Sequences with length greater than 300 were compared with the nr library and annotated. The mitochondrial genome map was constructed using OGDRAW (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html). RNA editing sites were predicted using the Plant Predictive RNA Editor (PREP) suite (http://prep.unl.edu/). Because the site has to stop the service, so we according to the algorithm and database to rewrite a tool PmtREP (http://112.86.217.82:9919/#/tool/alltool/detail/336), which is used to plant mitochondrial genome RNA editing sites prediction.
Analysis of RSCU and repeat sequences
Relative Synonymous Codon Usage (RSCU) is the ratio of the actual frequency of the use codon/theoretical frequency of the use codon. The RSCU of the mitochondrial genome of P. granatum was analyzed using a self-encoded Perl script, to screen for a unique CDS and determine the number of codons per gene. The blastn (v2.10.1) software was used to identify the repeat sequence of the genome itself. The -word_size was set to 7 and evalue was set to 1e-5, and the result was obtained after removing the redundancy.
Phylogenetic tree construction
The CDS was used to make the maximum likelihood evolutionary tree. MAFFT software (v7.427) was used for multi-sequence alignment of inter-species sequences. The matched sequences were connected from head to tail, and trimAl (v1.4.rev15) was used to trim. The jmodeltest-2.1.10 software was used to predict the model after trimming, and the model was determined as GTR type. The maximum likelihood evolutionary tree was constructed using RAxML v8.2.10 (https://cme.h-its.org/exelixis/software.html) software. The GTRGAMMA model and bootstrap = 1000 were selected as the parameter.
Genomic comparison analysis with related species
The Mafft software was used for global alignment of homologous gene sequences of different species, and dnasp5 was used to calculate the pi value of each gene. The comparative analysis of mitochondrial genome structure between P. granatum and proximal species was carried out using the CGVIEW (http://stothard.afns.ualberta.ca/cgview_server/) software. The nucmer (4.0.0beta2) software with maxmatch parameter was used for genome alignment between other sequences and assembly sequences, and Dot-plot was generated. The blastn (2.10.1+) software was used to compare the assembled species and the selected species in turn, and Multiple Synteny plots were drawn. The parameters were set as follows: -word_size was 7 and E-value was 1e-5. The blast software was used to find homologous sequences between chloroplasts and mitochondria genome, and the similarity was set to 70% with an E-value of 1E-5. The homologous fragments were visualized using circos v0.69-5 software.
Data availability
The raw sequencing data from the Illumina and Nanopore platforms generated during this study are available in GenBank. The associated BioProject and BioSample and SRA numbers are PRJNA971780 and SAMN35054861. SRA numbers of second and third generation data for the Illumina sequencing reads are SRR24519284 and SRR24554674, respectively. The mitogenome sequences were released into GenBank soon with the following accession numbers: OQ973289, OQ973290, OQ973291, OQ973292, OQ973293, OQ973294 and OQ973295. The plant samples are stored at the Shandong Institute of Pomology, Shandong, China (voucher numbers: SD-SL-026).
References
Chen L, Ren Y, Zhao J, Wang Y, Liu X, Zhao X, Yuan Z. Phylogenetic analysis of wild pomegranate (Punica granatum L.) based on its complete chloroplast genome from Tibet, China. Agronomy. 2023;13:126.
Shi J, Gao H, Wang S, Wu W, Tong R, Wang S, Li M, Jian Z, Wan R, Hu Q, et al. Exogenous arginine treatment maintains the appearance and nutraceutical properties of Hardand soft-seed pomegranates in cold storage. Front Nutr. 2022;9:828946.
Trainin T, Harel-Beja R, Bar-Ya’akov I, Ben-Simhon Z, Yahalomi R, Borochov-Neori H, Ophir R, Sherman M, Doron-Faigenboima A, Holland D. Fine mapping of the black peel color in pomegranate (Punica granatum L.) strongly suggests that a mutation in the anthocyanidin reductase (ANR) gene is responsible for the trait. Front Plant Sci. 2021;12:642019.
Venkitasamy C, Zhao LM, Zhang RH, Pan ZL. Pomegranate. In: Pan ZL, Zhang RH, Zicari S, editors. Integrated Processing Technologies for Food and Agricultural By-Products. Volume 2. Cambridge, MA, USA: Academic Press; 2019. pp. 181–216.
Ghasemi–Soloklui AA, Kordrostami M, Kordrostami A. Environmental and geographical conditions influence color, physical properties, and physiochemical composition of pomegranate fruits. Sci Rep. 2023;13:15447.
Anean HA, Mallasiy LO, Bader DMD, Shaat HA. Nano edible coatings and films combined with zinc oxide and pomegranate peel active phenol compounds has been to extend the shelf life of minimally processed pomegranates. Materials. 2023;16:1569.
Ökmen G, Giannetto D, Fazio F, Arslan K. Investigation of pomegranate (Punica granatum L.) flowers’ antioxidant properties and antibacterial activities against different staphylococcus species associated with bovine mastitis. Vet Sci. 2023;10:394.
Magangana TP, Makunga NP, Fawole OA, Stander MA, Opara UL. Antioxidant, antimicrobial, and metabolomic characterization of blanched pomegranate peel extracts: effect of cultivar. Molecules. 2022;27:2979.
Wang Q, Yuan T, Zhu X, Song G, Wang D, Li L, Huang M, Gong J. The phenolics, antioxidant activity and in vitro digestion of pomegranate (Punica granatum L.) peels: an investigation of steam Explosion pre-treatment. Front Nutr. 2023;10:1161970.
Li X, Zhe M, Huang Y, Fan W, Yang J, Zhu A. The evolution of mitochondrial genomes between two cymbidium sister species: dozens of circular chromosomes and the maintenance and deterioration of genome synteny. Genes. 2023;14:864.
Niu Y, Zhang T, Chen M, Chen G, Liu Z, Yu R, Han X, Chen K, Huang A, Chen C, et al. Analysis of the complete mitochondrial genome of the bitter gourd (Momordica charantia). Plants. 2023;12:1686.
Lu C, Gao LZ, Zhang QJ. A high-quality genome assembly of the mitochondrial genome of the oil-tea tree Camellia gigantocarpa (Theaceae). Diversity. 2022;14:850.
Zhang N, Li Y, Halanych KM, Kong L. Li QA comparative analysis of mitochondrial ORFs provides new insights on expansion of mitochondrial genome size in Arcidae. BMC Genomics. 2022;23:809.
You C, Cui T, Zhang C, Zang S, Su Y, Que Y. Assembly of the complete mitochondrial genome of Gelsemium Elegans revealed the existence of homologous conformations generated by a repeat mediated recombination. Int J Mol Sci. 2023;24:527.
Li J, Li J, Ma Y, Kou L, Wei J, Wang W. The complete mitochondrial genome of okra (Abelmoschus esculentus): using nanopore long reads to investigate gene transfer from chloroplast genomes and rearrangements of mitochondrial DNA molecules. BMC Genomics. 2022;23:481.
Bi C, Lu N, Xu Y, He C. Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. Int J Mol Sci. 2020;21:3778.
Ma Q, Wang Y, Li S, Wen J, Zhu L, Yan K, Du Y, Ren J, Li S, Chen Z, et al. Assembly and comparative analysis of the first complete mitochondrial genome of Acer Truncatum Bunge: a woody oil-tree species producing nervonic acid. BMC Plant Biol. 2022;22:29.
Wu ZQ, Liao XZ, Zhang XN, Tembrock LR, Broz A. Genomic architectural variation of plant mitochondria-A review of multichromosomal structuring. J Syst Evol. 2022;60:160–8.
Tsujimura M, Kaneko T, Sakamoto T, Kimura S, Shigyo M, Yamagishi H, Terachi T. Multichromosomal structure of the onion mitochondrial genome and a transcript analysis. Mitochondrion. 2019;46:179–86.
Wang Y, Chen S, Chen J, Chen C, Lin X, Peng H, Zhao Q, Wang X. Characterization and phylogenetic analysis of the complete mitochondrial genome sequence of Photinia serratifolia. Sci Rep. 2023;13:770.
Niu Y, Gao C, Liu J. Complete mitochondrial genomes of three Mangifera species, their genomic structure and gene transfer from chloroplast genomes. BMC Genomics. 2022;23:147.
Pinard D, Myburg AA, Mizrachi E. The plastid and mitochondrial genomes of Eucalyptus grandis. BMC Genomics. 2019;20:132.
Goremykin VV, Lockhart PJ, Viola R, Velasco R. The mitochondrial genome of malus domestica and the import-driven hypothesis of mitochondrial genome expansion in seed plants. Plant J. 2012;71:615–26.
Zhang S, Yin Z, Wu X, Li C, Xie K, Deng X, Grosser JW, Guo W. Assembly of Satsuma mandarin mitochondrial genome and identification of cytoplasmic male sterility–specific ORFs in a somatic cybrid of pummelo. Tree Genet Genomes. 2020;16:84.
Wang S, Li D, Yao X, Song Q, Wang Z, Zhang Q, Zhong C, Liu Y, Huang H. Evolution and diversification of kiwifruit mitogenomes through extensive whole-genome rearrangement and mosaic loss of intergenic sequences in a highly variable region. Genome Biol Evol. 2019;11(4):1192–206.
Lu G, Zhang K, Que Y, Li Y. Assembly and analysis of the first complete mitochondrial genome of Punica granatum and the gene transfer from chloroplast genome. Front Plant Sci. 2023;14:1132551.
Ke SJ, Liu DK, Tu XD, He X, Zhang MM, Zhu MJ, Zhang DY, Zhang CL, Lan SR, Liu ZJ. Apostasia mitochondrial genome analysis and monocot mitochondria phylogenomics. Int J Mol Sci. 2023;24:7837.
Guo Y, Li Z, Jin S, Chen S, Li F, Wu H. Assembly and comparative analysis of the complete mitochondrial genome of two species of Calla lilies (Zantedeschia, Araceae). Int J Mol Sci. 2023;24:9566.
Putintseva YA, Bondar EI, Simonov EP, Sharov VV, Oreshkova NV, Kuzmin DA, Konstantinov YM, Shmakov VN, Belkov VI, Sadovsky MG, et al. Siberian larch (Larix sibirica Ledeb.) Mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genom. 2020;21:654.
Yu X, Duan Z, Wang Y, Zhang Q, Li W. Sequence analysis of the complete mitochondrial genome of a medicinal plant, Vitex rotundifolia Linnaeus f. (Lamiales: Lamiaceae). Genes. 2022;13:839.
Cao P, Huang Y, Zong M, Xu Z. De Novo Assembly and comparative analysis of the complete mitochondrial genome of Chaenomeles speciosa (Sweet) Nakai revealed the existence of two structuralisomers. Genes. 2023;14:526.
Yang JX, Dierckxsens N, Bai MZ, Guo YY. Multichromosomal mitochondrial genome of Paphiopedilum micranthum: compact and fragmented genome, and rampant intracellular gene transfer. Int J Mol Sci. 2023;24:3976.
Liu D, Guo H, Zhu J, Qu K, Chen Y, Guo Y, Ding P, Yang H, Xu T, Jing Q, et al. Complex physical structure of complete mitochondrial genome of Quercus acutissima (Fagaceae): a significant energy plant. Genes. 2022;13:1321.
Qiao Y, Zhang X, Li Z, Song Y, Sun Z. Assembly and comparative analysis of the complete mitochondrial genome of bupleurum chinense DC. BMC Genomics. 2022;23:664.
Terasawa K, Odahara M, Kabeya Y, Kikugawa T, Sekine Y, Fujiwara M, Sato N. The mitochondrial genome of the moss Physcomitrella patens sheds new light on mitochondrial evolution in land plants. Mol Biol Evol. 2007;24:699–709.
Guo W, Zhu A, Fan W, Adams RP, Mower JP. Extensive shifts from cis- to trans-splicing of Gymnosperm mitochondrial introns. Mol Biol Evol. 2020;37:1615–20.
Yang J, Wariss H, Tao L, Zhang R, Yun Q, Hollingsworth P, Dao Z, Luo G, Guo H, Ma Y, Sun W. De novo genome assembly of the endangered Acer Yangbiense, a plant species with extremely small populations endemic to Yunnan Province, China. Gigascience. 2019;8(7):giz085.
Ye N, Wang X, Li J, Bi C. Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant Salix suchowensis. PeerJ. 2017;5(1):e3148.
Shi S, Liu Y, Xia R, Qin L. Comprehensive analysis of codon usage in Quercus chloroplast genome and focus on psbA gene. Genes. 2022;13(11):2156.
Zeng Y, Shen L, Chen S, Qu S, Hou N. Codon usage profiling of chloroplast genome in Juglandaceae. Forests. 2023;14(2):378.
Wu P, Xiao W, Luo Y, Xiong Z, Chen X, He J, Sha A, Gui M, Li Q. Comprehensive analysis of codon bias in 13 Ganoderma mitochondrial genomes. Front Microbiol. 2023;14:1170790.
Li J, Tang H, Luo H, Tang J, Zhong N, Xiao L. Complete mitochondrial genome assembly and comparison of Camellia sinensis var. Assamica Cv. Duntsa Front Plant Sci. 2023;14:1117002.
Liu D, Qu K, Yuan Y, Zhao Z, Chen Y, Han B, Li W, El-Kassaby YA, Yin Y, Xie X, Tong B, Liu H. Complete sequence and comparative analysis of the mitochondrial genome of the rare and endangered Clematis acerifolia, the first clematis mitogenome to provide new insights into the phylogenetic evolutionary status of the genus. Front Genet. 2023;13:1050040.
Guo W, Felix G, Fan W, Young G. Ginkgo and Welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol Biol Evol. 2016;33(6):1448–60.
Xiong Y, Lei X, Bai S, Xiong Y, Liu W, Wu W, Yu Q, Dong Z, Yang J, Ma X. Genomic survey sequencing, development and characterization of single- and multi-locus genomic SSR markers of Elymus sibiricus L. BMC Plant Biol. 2021;21:3.
Li J, Xu Y, Shan Y, Pei X, Yong S, Liu C, Yu J. Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat–mediated recombination. Planta. 2021;254:36.
Dong S, Zhao C, Chen F, Liu Y, Zhang S, Wu H, Zhang L, Liu Y. The complete mitochondrial genome of the early flowering plant nymphaea colorata is highly repetitive with low recombination. BMC Genomics. 2018;19:614.
Shan Y, Li J, Zhang X, Yu J. The complete mitochondrial genome of Amorphophallus Albus and development of molecular markers for five Amorphophallus species based on mitochondrial DNA. Front. Plant Sci. 2023;14:1180417.
Sheng W, Deng J, Wang C, Kuang Q. The garden asparagus (Asparagus officinalis L.) mitochondrial genome revealed rich sequence variation throughout whole sequencing data. Front Plant Sci. 2023;14:1140043.
Habib S, Dong S, Liu Y, Liao W, Zhang S. The complete mitochondrial genome of Cycas debaoensis revealed unexpected static evolution in gymnosperm species. PLoS ONE. 2021;16(7):e0255091.
He P, Xiao GH, Liu H, Zhang LH, Zhao L, Tang MJ, Huang S, An YJ, Yu JN. Two pivotal RNA editing sites in the mitochondrial atp1 mRNA are required for ATP synthase to produce sufficient ATP for cotton fiber cell elongation. New Phytol. 2018;218:167–82.
Shtratnikova VY, Schelkunov MI, Penin AA, Logacheva MD. Mitochondrial genome of the nonphotosynthetic mycoheterotrophic plant Hypopitys monotropa, its structure, gene expression and RNA editing. PeerJ. 2020;8:e9309.
Gerke P, Szovenyi P, Neubauer A, Lenz H, Gutmann B, McDowell R, Small I, Schallenberg-Rudinger M, Knoop V. Towards a plant model for enigmatic U-to-C RNA editing: the organelle genomes, transcriptomes, editomes and candidate RNA editing factors in the hornwort Anthoceros agrestis. New Phytol. 2020;225:1974–92.
Mower JP, Palmer JD. Patterns of partial RNA editing in mitochondrial genes of Beta vulgaris. Mol Genet Genom. 2006;276:285–93.
Yang H, Chen H, Ni Y, Li J, Cai Y, Wang J, Liu C. Mitochondrial genome sequence of Salvia officinalis (Lamiales: Lamiaceae) suggests diverse genome structures in cogeneric species and finds the stop gain of genes through RNA editing events. Int J Mol Sci. 2023;24:5372.
Kovar L, Nageswara-Rao M, Ortega-Rodriguez S, Dugas DV, Straub S, Cronn R, Strickler SR, Hughes CE, Hanley KA, Rodriguez DN, et al. PacBio-based mitochondrial genome assembly of Leucaena Trichandra (Leguminosae) and an intrageneric assessment of mitochondrial RNA editing. Genome Biol Evol. 2018;10(9):2501–17.
Zhang X, Shan Y, Li J, Qin Q, Yu J, Deng H. Assembly of the complete mitochondrial genome of Pereskia aculeata revealed that two pairs of repetitive elements mediated the recombination of the genome. Int J Mol Sci. 2023;24:8366.
Hong Z, Liao XZ, Ye YJ, Zhang NN, Yang ZJ, Zhu WD, Gao W, Sharbrough J, Tembrock LR, Xu DP, et al. A complete mitochondrial genome for fragrant Chinese rosewood (Dalbergia Odorifera, Fabaceae) with comparative analyses of genome structure and intergenomic sequence transfers. BMC Genom. 2021;22:672.
Usha T, Middha SK, Babu D, Goyal AK, Das AJ, Saini D, Sarangi A, Krishnamurthy V, Prasannakumar MK, Saini DK, Sidhalinghamurthy KR. Hybrid assembly and annotation of the genome of the Indian Punica granatum, a superfood. Front Genet. 2022;13:786825.
Cole LW, Guo W, Mower JP, Palmer JD. High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol Biol Evol. 2018;35(11):2773–85.
Gui ST, Wu ZH, Zhang HY, Zheng YZ, Zhu ZX, Liang DQ, Ding Y. The mitochondrial genome map of Nelumbo nucifera reveals ancient evolutionary features. Sci Rep. 2016;6:11.
Nguyen VB, Giang VNL, Waminal NE, Park HS, Kim NH, Jang W, Lee J, Yang T. Comprehensive comparative analysis of chloroplast genomes from seven Panax species and development of an authentication system based on species-unique single nucleotide polymorphism markers. J Ginseng Res. 2020;44(1):135–44.
Kitazaki K, Kubo T, Kagami H, Matsumoto T, Fujita A, Matsuhira H, Matsunaga M, Mikami T. A horizontally transferred Trna (Cys) gene in the sugar beet mitochondrial genome: evidence that the gene is present in diverse angiosperms and its transcript is aminoacylated. Plant J Cell Mol Biol. 2011;68:262–72.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–100.
Koren S, Walenz BP, Berlin K, Miller JR, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36.
Chan PP, Lowe TM. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol Biol. 2019; 1962:1–14.
Acknowledgements
Not applicable.
Funding
This work was supported by the Shandong Key Research and Development Projects (2022TZXD009, 2021LZGC007), Agricultural Science and Technology Innovation Engineering Discipline Team of Shandong Academy of Agricultural Sciences (CXGC2022D02, CXGC2023A12).
Author information
Authors and Affiliations
Contributions
Y.Y.: Conceptualization. F.L. and W.C.: analyzed the data, F.L. and Y.X.: wrote original manuscript. W.Z., A. M. and Y.Y.: reviewed and edited the manuscript. F.L., W.C. and Y.Y.: methodology. F.L. and Y.Y.: project administration. Y.Y.: funding acquisition. All authors reviewed the manuscript and agreed to the submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All plant materials used in this study were grown and collected according to institutional and national guidelines. P. granatum ‘Taishanhong’ was a common cultivar and was obtained commercially. The study, including plant samples, complies with relevant institutional, national, and international guidelines and legislation. No specific permits were required for plant collection.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
. Branched conformation of P. granatum mitogenome. Fig. S2. Data analysis depth chart of P. granatum mitogenome. Supplementary Tables: Table S1. General features of the P. granatum mitogenome. Table S2. Codon usage of P. granatum mitogenome. Table S3. Repeated sequence analysis of P. granatum mitogenome. Table S4. The RNA editing events prediction in P. granatum mitogenome. Table S5. The nucleotide variability of P. granatum mitogenome. Table S6. Comparison information of chloroplast and mitochondrial genome in P. granatum. Table S7. Gene sequence of chloroplast transfer to mitochondria in P. granatum.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Feng, L., Wang, Z., Wang, C. et al. Multichromosomal mitochondrial genome of Punica granatum: comparative evolutionary analysis and gene transformation from chloroplast genomes. BMC Plant Biol 23, 512 (2023). https://doi.org/10.1186/s12870-023-04538-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04538-8