
Abstract
Background
Wheat (Triticum aestivum L.) is a major cereal crop that is grown worldwide, and it is highly dependent on sufficient N supply. The molecular mechanisms associated with nitrate uptake and assimilation are still poorly understood in wheat. In plants, NRT2 family proteins play a crucial role in NO3– acquisition and translocation under nitrate limited conditions. However, the biological functions of these genes in wheat are still unclear, especially their roles in NO3– uptake and assimilation.
Results
In this study, a comprehensive analysis of wheat TaNRT2 genes was conducted using bioinformatics and molecular biology methods, and 49 TaNRT2 genes were identified. A phylogenetic analysis clustered the TaNRT2 genes into three clades. The genes that clustered on the same phylogenetic branch had similar gene structures and nitrate assimilation functions. The identified genes were further mapped onto the 13 wheat chromosomes, and the results showed that a large duplication event had occurred on chromosome 6. To explore the TaNRT2 gene expression profiles in wheat, we performed transcriptome sequencing after low nitrate treatment for three days. Transcriptome analysis revealed the expression levels of all TaNRT2 genes in shoots and roots, and based on the expression profiles, three highly expressed genes (TaNRT2-6A.2, TaNRT2-6A.6, and TaNRT2-6B.4) were selected for qPCR analysis in two different wheat cultivars (‘Mianmai367’ and ‘Nanmai660’) under nitrate-limited and normal conditions. All three genes were upregulated under nitrate-limited conditions and highly expressed in the high nitrogen use efficiency (NUE) wheat ‘Mianmai367’ under low nitrate conditions.
Conclusion
We systematically identified 49 NRT2 genes in wheat and analysed the transcript levels of all TaNRT2s under nitrate deficient conditions and over the whole growth period. The results suggest that these genes play important roles in nitrate absorption, distribution, and accumulation. This study provides valuable information and key candidate genes for further studies on the function of TaNRT2s in wheat.
Similar content being viewed by others

Introduction
Nitrogen (N) is the second most important crop input factor after water. It is also an important component of most biomacromolecules and many secondary and signaling compounds in plants, such as proteins, nucleic acids, cell wall components, phytohormones, and vitamins [1, 2]. Therefore, nitrogen deficiency could severely limit plant growth and development. This is particularly true for wheat. Wheat plants do not establish symbiotic associations with N2-fixing microbes [3]. Therefore, chemical fertilizers have historically been used to maintain or increase crop yields. However, these chemical fertilizers have been mismanaged, resulting in environmental pollution and decreased nutrient-use efficiency (NUE) [4]. For example, only one-third of the applied nitrogen is utilized by wheat, which suggests that there is potential for increasing its NUE [5]. The remaining N is released into the environment through leaching and volatilization [6]. This means that low wheat NUE and excess N fertilizer applications are aggravate environmental pollution and cause ecological deterioration [7, 8]. Therefore, improving the NUE will improve the sustainability of wheat production. However, achieving greater NUE is challenged by the complexity of the trait, which is comprised of processes associated with nitrogen uptake, transport, reduction, assimilation, translocation, and remobilization.
Nitrogen is available to plant roots in several different forms, such as NO3−, NH4+, and organic molecules, such as amino acids [9]. Nitrate is one of the most important N sources for plants. Nitrogen uptake is the first step in nitrate assimilation and can be manipulated to enhance NUE. Plants have evolved regulated, energy-dependent systems for the uptake of NO3− that use both high- and low-affinity transporters. The nitrate transporter 1 (NRT1)/peptide transporter (PTR) family (NPF), NRT2 family, chloride channel (CLC) family, and slow anion channel (SLAC) protein family are the four protein families that play key roles in NO3− transport [10, 11]. The NRT1 and NRT2 families have been identified as being involved in low-affinity nitrate transporter systems (LATSs) and high-affinity nitrate transporter systems (HATSs), respectively. The LATS is activated when nitrate concentrations are high (> 1 mM), whereas the HATS is activated when nitrate concentrations are low (< 1 mM) [12, 13]. The NRT2s, which are thought to be involved in the major transporter system responsible for nitrate uptake in plants, are membrane associated proteins and contribute specifically to nitrate-inducible steps.
The first NRT2 family transporters were discovered in a chlorate-resistant mutant (crnA) of Aspergillus nidulans [14, 15]. Subsequently, numerous studies have investigated the functional roles of the plant NRT2 family and important progress has been made. There are 7 NRT2 genes in Arabidopsis [10, 16], 4 in rice [17], 4 in maize [18], 31 in rapeseed [19, 20], 13 in poplar [21], 4 in tomato [22], and 5 in wild soybean (Glycine soja) [23]. In Arabidopsis, four AtNRT2 transporters (AtNRT2.1, AtNRT2.2, AtNRT2.3, and AtNRT2.4) are involved in nitrate uptake. The AtNRT2.1 and AtNRT2.2 genes play key roles in the regulation of high-affinity NO3– uptake and nrt2.1nrt2.2 reduces the inducible high-affinity transport system (IHATS) by up to 80% in Arabidopsis thaliana [24, 25]. AtNRT2.4 has a role in both the roots and shoots under N starvation [26] and AtNRT2.5 is the most abundant transcript in adult plants among the seven AtNRT2 family members after long-term nitrogen starvation [27]. Furthermore, AtNRT2.7 is specifically highly expressed in reproductive organs, reaching a maximum in dry seeds, and AtNRT2.7 is the only NRT2 transporter located in the tonoplast [28].
In crops, the homologs of AtNRT2s have been shown to perform numerous roles in N uptake, transport, and utilization processes across all developmental stages. In rice, OsNRT2.1 and OsNRT2.2 share the same coding sequences (CDSs) with different 5′- and 3′-untranslated regions (UTRs) and have high similarities with maize ZmNRT2 genes, while OsNRT2.3 is more closely related to AtNRT2.5, and OsNRT2.4 is more closely related to AtNRT2.7 [17]. OsNRT2.3 mRNA has been previously spliced into OsNRT2.3a and OsNRT2.3b [29]. OsNRT2.3a plays a key role in long-distance nitrate transport from root to shoot at low nitrate supply levels [30], OsNRT2.3b plays a critical role in sensing the cytosolic pH of phloem cells and increased OsNRT2.3b expression improves grain yield and NUE [31]. OsNRT2.4 has been shown to be a dualaffinity nitrate transporter and is required for nitrate-regulated root and shoot growth [32]. In wheat, TaNRT2.5 is expressed in the root, leaf, embryo, and shell and can increase seed vigour, grain nitrate accumulation, and yield [33]. In maize, only ZmNRT2.1 plays a role in nitrate uptake along the root axis [34]. In summary, NRT2 homologs play key roles in nitrate uptake and utilization in plants.
Wheat (Triticum aestivum L.) is one of the three main cereal crops across the globe. The ability to uptake N is heavily dependent on the functional efficiency of the nitrate transporter, which is genetically determined in many crops. However, TaNRT2 family members have not been systematically identified, and their expression has been analysed under nitrate deficiency conditions in wheat. This is due to the complexity of its genome. In this study, a genome-wide identification of TaNRT2 members in wheat was performed. The gene structures, chromosomal locations, cis-elements, and conserved motifs of all TaNRT2s were also analysed. Furthermore, a transcriptome analysis of all TaNRT2s was conducted under nitrate starvation conditions. This study reveals the characteristics of NRT2 genes in wheat and provides valuable information and candidate gene resources for future functional analyses that could be used to genetically improve the NUE of wheat.
Results
Identification of the NRT2 gene family in wheat
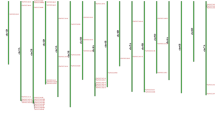
To identify the NRT2 gene family in wheat, whole-genome scanning and a Blastp search were used to identify the genes that contained the conserved domain (MFS). A total of 49 NRT2 genes were identified in the wheat genome. These consisted of 46 high-confidence genes and three low-confidence genes (Table 1). The 49 NRT2 genes were unevenly distributed on the 13 wheat chromosomes and 38 of them were located on chromosome 6 (Fig. 1). The TaNRT2s on chromosome 6 showed multiple duplication to form tandemly duplicated gene clusters. There was also good collinearity among the 6A, 6B, and 6D homologous genes (Fig. 1). In addition, the characteristics of the TaNRT2 genes, including the CDS length, protein length, molecular weight (MW), isoelectric point (pI), and predicted subcellular localization, were systematically evaluated (Table 1). The CDS lengths of the TaNRT2 genes ranged from 780 (TaNRT2-U.2) to 1698 (TaNRT2-6B.5) and the corresponding protein lengths ranged from 259 to 565. The protein MWs ranged from 28.00 kDa (TaNRT2-U.2) to 60.98 kDa (TaNRT2-6B.5) and the average pIs of the TaNRT2 proteins ranged from 7.51 (TaNRT2-7D) to 9.77 (TaNRT2-7A). The subcellular localization prediction for TaNRT2 proteins suggested that most TaNRT2s were located on the plasma membrane.

Chromosomal localization and collinearity of TaNRT2 genes on bread wheat genome. The black line between the gene names indicated that they were tandem repeat gene pairs. Gene locations are shown by the scale. The gene location on each chromosome is represented by grey lines. The collinear relationships of TaNRT2 genes are indicated by blue dotted lines
Phylogenetic analysis of TaNRT2s
To investigate the phylogenetic relationship between TaNRT2s and NRT2s from other plant species, a neighbor-joining phylogenetic tree consisting of 49 TaNRT2s, 4 OsNRT2s, 4 ZmNRT2s, and 7 AtNRT2s was generated after multi-alignment of the protein sequences (Fig. 2). The total number of NRT2 genes in wheat is far larger than rice, maize and Arabidopsis, which is partly a result of the hexaploidy nature of wheat. However, even when corrected for ploidy level, the number of NRT2 genes in the wheat ABD sub-genome was significantly larger than that in rice and Arabidopsis (Fig. S1a). The ratio of total NRT2 genes between wheat and rice or wheat and Arabidopsis was significantly higher than the expected 3:1 ratio (Fig. S1b). This indicated that the expansion of NRT2 genes in wheat was not only due to hexaploidy but also due to a large number of tandem duplications during the evolution of wheat. According to the phylogenetic tree, the NRT2s were clustered into three main clades and each clade contained monocots and dicots. Clade 1 contained the most members, including 41 TaNRT2s, 5 AtNRT2s, 3 ZmNRT2s and 2 OsNRT2s. Of the 41 TaNRT2s in clade 1, 36 were located on chromosome 6 and divided into two branches, suggesting that two gene duplication events occurred on chromosome 6 during the formation and evolution of the wheat NRT2 gene. The five TaNRT2s in clade 2 and the three TaNRT2s in clade 3 were homologous to AtNRT2.5 and AtNRT2.7, respectively. These results suggest that duplications and multiplications have contributed to the expansion of the TaNRT2 gene family in wheat.

Phylogenetic tree of the NRT2 genes family. Phylogenetic tree of NRT2 members in higher plants was generated by protein sequence alignment with MEGA 6.0 using the neighbor-joining method, displayed by Evolview 2.0. All NRT2 proteins were assigned into three groups as clade1, 2 and 3 (indicated by red, green and blue, respectively). At: A. thaliana, Os: O. sativa, Zm: Z. mays, Ta: T. aestivum (marked by a light green, blue, yellow and pink circle, respectively)
Conserved domain and gene structure analysis
The conserved protein motifs, conserved domain, and gene structure were characterized to further understand the evolutionary characteristics of the TaNRT2 gene family. Ten motifs were identified using MEME to illustrate the protein structure of the TaNRT2 family (Fig. 3a and b). The results showed that 37 TaNRT2s contained all the motifs, seven TaNRT2s contained nine motifs and three TaNRT2s contained eight motifs. Only motif 1 was present in all 49 TaNRT2 proteins. TaNRT2-U.2 contained the fewest number of motifs because it contained only motifs 1, 4, 5, and 7. The TaNRT2 gene family was identified by the presence of a nitrate transmembrane transporter domain (Pfam PLN00028).

The gene structure analysis and conserved motifs of TaNRT2 genes. a Conserved motifs, conserved domain and gene structures of TaNRT2 genes. MEME motif: Ten MEME motifs are colored by different color. The length of each box in the figure does not represent the actual motif size. NCBI CDD: The conserved domains are represented by purple boxes. Gene structure: exons, introns, and untranslated regions (UTRs) are indicated by yellow rectangles, gray lines, and green rectangles, respectively. b Sequence logo conserved motif of TaNRT2 proteins. The overall height of each stack represents the degree of conservation at this position, while the height of individual letters within each stack indicates the relative frequency of the corresponding amino acids
The exon–intron structures were analysed to further understand the structural characteristics of the TaNRT2 genes (Fig. 3a). The results showed that most TaNRT2 genes had similar gene structures. There were 1–2 exons in the TaNRT2 genes. Among the 49 TaNRT2 genes, four genes (TaNRT2-7A, TaNRT2-7B, TaNRT2-7D, and TaNRT2-6B.6) contained one intron, while the other 45 TaNRT2 genes had no intron. These results suggested that the similar features of wheat NRT2 genes might be due to duplication events during species evolution.
Prediction of cis‑regulatory elements (CARE) in TaNRT2s
Cis-regulatory elements play a role in the transcriptional regulation of various biological processes, including phytohormone responses, defence responses, and developmental processes. To further understand the potential regulatory mechanism controlling TaNRT2 genes, the PlantCARE database (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/) was used to identify putative cis-acting elements in the 2000 bp promoter region of TaNRT2s [35]. A total of 16 CAREs were identified in the 49 TaNRT2 genes, including hormone responses, defence and stress-responsive, light response, growth, and development regulation (Fig. 4a). The CAREs involved in light, MeJA, abscisic acid response, and anaerobic induction were the most abundant in the TaNRT2 gene family (Fig. 4b). These results suggest that TaNRT2 family members may play roles in diverse developmental processes, such as phytohormones, stress responses, and light responses.

Cis-acting regulatory elements (CAREs) of the TaNRT2 gene family. a The CAREs analysis was performed with a 2 kb upstream region using PlantCARE online server. b The distribution of CAREs in the promoter of TaNRT2 genes. Most commonly occurring CAREs in TaNRT2s
Expression profiling of TaNRT2s in various tissues
The publicly accessible RNA seq database for hexaploidy common wheat (var. Chinese spring), which includes various tissues and stages, was explored in order to analyse the expression profiles of TaNRT2 genes in different tissues, such as roots, stems, leaves, spikes, and grains. The transcripts per million (TPM) values for the TaNRT2 genes were used to construct a heatmap (Fig. 5). The heatmap indicated that most TaNRT2 genes were specifically expressed in the roots, especially in the roots during the Z13 and Z39 stages. However, three genes (TaNRT2-3A, TaNRT2-3B, and TaNRT2-3D) also showed high expression levels in Z85 grains, which suggested that these three genes may participate in nitrogen accumulation in grains. Furthermore, TaNRT2-7A, TaNRT2-7B, and TaNRT2-7D were highly expressed in the leaves, which indicated that these genes may play important roles in nitrate distribution. The expression pattern for TaNRT2 genes suggested that the wheat NRT2 family members can be divided into two groups. The genes in group I showed low expression or were not expressed in the roots and other tissues. However, the group II genes showed highly specific expression in the roots. These results showed that these genes play important roles in NO3− absorption and transportation under N-limiting conditions.

Heatmap representing the expression pattern of TaNRT2 genes in various developmental stages. The TPM values normalized by logarithmic scale were used to construct the heatmap. Z10 ~ Z85 represent different growth stage of wheat. Different colors represent relative expression levels, as shown in the legend on the right. The horizontal axis represents the names and classifications genes, and the vertical axis represents various tissues. The rows of the heat map are clustered according to the expression patterns
RNA-seq analysis of nitrogen deficiency
To explore whether TaNRT2 genes are induced by nitrogen deficiency, the widely cultivated variety ‘Chuanmai104’ was used for the nitrogen deficiency treatment. When grown to the two-leaf stage, one group of seedlings was subjected to three days of low nitrogen (0 mM nitrate) and seedlings grown in normal nutrient solution (5 mM nitrate) were used as controls. After treatment, the seedlings were divided into roots and shoots. Three biological replicates and a total of 12 samples were used for the RNA-seq analysis. The differentially expressed gene (DEG) analysis indicated that there were 4944 significantly upregulated genes and 3458 significantly downregulated genes in the roots after low nitrogen treatment (Fig. 6a). However, the number of DEGs substantially decreased in the shoots. There were only 516 significantly upregulated genes and 1054 significantly downregulated genes in the low-nitrogen treated shoots (Fig. 6b). This indicated that there are more genes responding to nitrogen signals in the roots.

KEGG enrichment scatter plot of root and volcano plot under nitrogen deficiency treatment. The volcano indicating the DEGs in roots (a) and shoots (b), each dot in the figure represents a particular gene, and the red dots indicate significantly up-regulated genes, the green dots indicate significantly down-regulated genes, and the blue dots represent non-significant differential genes. The 20 most significantly DEG-enriched pathways of wheat seedling in roots (c) and shoots (d) under nitrate deficiency treatment
GO enrichment analysis revealed that carbohydrate and polysaccharide metabolic processes were enriched in the roots, while photosynthesis and oxidoreductase activity were enriched in the shoots (Fig. S2). KEGG analysis showed that nitrogen metabolism and glutathione metabolism were the two most significantly altered pathways in the roots after the plants had been subjected to the nitrogen deficiency treatment (Fig. 6c), while photosynthesis-related pathways and nitrogen metabolism were significantly enriched in the shoots (Fig. 6d). These results suggest that genes in the roots and shoots respond to stress caused by nitrogen deficiency.
To investigate the expression of the TaNRT2 genes, the FPKM values of all the TaNRT2 genes identified by RNA-seq were used to construct a heatmap (Fig. 7). The results showed that the expression of 24 TaNRT2 genes were induced by nitrogen deficiency. Among them, TaNRT2-6D.1 and TaNRT2-6A.6 were most significantly induced by nitrogen deficiency in the roots, indicating that they may play important roles in nitrogen absorption and metabolism. In addition, the expression of TaNRT2-7A/B/D were upregulated in both the roots and shoots after the nitrogen deficiency treatment. This indicates that TaNRT2-7A/B/D may also participate in the nitrogen transfer and accumulation in the shoots in addition to nitrogen uptake in the roots. The expression of the remaining 25 TaNRT2 genes did not change under nitrogen deficiency condition, indicating that the genes in the TaNRT2 family might have functionally differentiated or been made functionally redundant during evolution.

Heatmap representing the expression of TaNRT2 genes under nitrogen deficiency treatment. The FPKM values of all TaNRT2s from transcriptome databases were used to construct the heatmap. The color represent relative expression levels. T_R: low nitrogen treated roots, CK_R: control roots, T_S: low nitrogen treated shoot, CK_S: control shoots
Phenotypic and expression analyses of two different wheat varieties
Two different NUE wheat varieties, ‘Mianmai367’ and ‘Nanmai660’, were used to investigate the phenotypes under nitrate-limited conditions. The two-leaf stage seedlings were transferred to high nitrate (5 mM, HN) and low nitrate (0.1 mM, LN) Hoagland hydroponic solutions as the control and treatment conditions, respectively. After treatment for 12 days, the nitrate-deficient phenotypes of the two varieties were evaluated and the dry weight and nitrogen content were measured. The cultivar ‘Mianmai367’ showed obvious tolerance to low nitrate stress compared to ‘Nanmai660’ (Fig. 8a). The biomass results were consistent with the observed phenotypes. The root and shoot biomasses for ‘Nanmai660’ were obviously lower than those for ‘Mianmai367’ (Fig. 8b). The nitrogen content in ‘Nanmai660’ was also lower than in ‘Mianmai367’ (Fig. 8c). The results showed that there was a significant difference in LN tolerance between the two wheat genotypes, further indicating that the nitrogen nutritional activities of wheat are genetically regulated through gene expression that increases its efficient use or tolerance to low nitrogen.

Phenotype comparison between two wheat cultivars under low nitrogen condition. a Phenotype of ‘Mianmai367’ and ‘Nanmai660’ under HN (5 mM NO3−) and LN (0.1 mM NO3−) conditions for 12 days; b Dry weight of shoot and root under HN and LN condition; c Nitrogen contents of shoot and root under HN and LN condition. White scale bar represents 10 cm. Data are means ± SEM (n = 4). Differences between mean values of treatments and controls were compared using t—tests (* P < 0.05, ** P < 0.01). S: shoot; R: root; d-f Relative expression of three NRT2 genes in root of two different wheat cultivars under phenotype condition
To further understand the NUE differences between the two wheat varieties, we further investigated several highly expressed genes (TaNRT2-6A.2, TaNRT2-6A.6, and TaNRT2-6B.4) under low nitrate conditions. The results showed that the expression of the three genes were all upregulated under N-limiting conditions in the two varieties. However, the three genes had higher expression levels in ‘Mianmai367’ than in ‘Nanmai660’ (Fig. 8d-f). These results showed that ‘Mianmai367’ had a greater ability to efficiently utilize N than ‘Nanmai660’.
Discussion
In the past two decades, the NRT2 gene family has been identified in numerous plant genomes, such as Arabidopsis [10, 16], rice [17], maize [18], rapeseed [19], poplar [21] and tomato [22], and the number of NRT2 genes ranges from 4 (rice, maize and tomato) to 31 (rapeseed). In this study, a genome-wide analysis revealed 49 NRT2 members in wheat and identified three new genes compared to a previous report [36]. The numbers of wheat NRT2 genes were significantly more than those of rice, Arabidopsis and other species at both the genome and sub-genome level (Fig. S1). According to the evolutionary relationship, these genes can be divided into three clades, which is consistent with reports in other species. In addition, all the NRT2 genes had a conserved MFS domain and multiple transmembrane domains (Table 1).
A total of 38 out of the 49 TaNRT2 genes were located on chromosome 6 (Fig. 1), and there were three tandem repeat gene pairs on chromosome 6A, 6B and 6D. These genes also had good collinearity between ABD sub-genomes and a close evolutionary relationship (Fig. 2). It is speculated that whole genome duplication and tandem duplication might contribute to NRT2 gene expansion in wheat. It has been reported that 16 gene duplication events occurred during the evolution of the wheat NRT2 gene family [37]. All TaNRT2 genes on chromosome 2 and 6 were classified into clade 1, since an ancient NRT2 gene duplicated into two copies on chromosomes 2 and 6 after the monocot-dicot split [37].
Many studies have indicated that the exon–intron patterns are commonly conserved in gene families or subfamilies in plants. In this study, we analysed the gene location, gene structure, conserved motifs, cis-acting regulatory elements, and gene expression profiles of all the TaNRT2 genes (Figs. 3, 4 and 5). The gene structure analysis showed that all the TaNRT2s had one exon, except for the three genes on chromosome 7 and one gene on chromosome 6 which had two exons (Fig. 3a). The intron length of genes on chromosome 7 (TaNRT2-7A/7B/7D), especially TaNRT2-7D, was even longer than the coding region. Furthermore, a previous study revealed that TaNRT2-7A/7B/7D had experienced a third duplication during the evolution of the wheat NRT2 gene family [37]. This suggests that an extra exon acquisition might have occurred in TaNRT2-7A/7B/7D during wheat evolution and that this has led to the various structures seen in this family.
The NRT2 family is involved in the high-affinity nitrate transporter systems and plays vital roles in both nitrate uptake and translocation in plants. We conducted a transcriptome analysis of bread wheat under nitrate deficiency conditions to further understand the TaNRT2 transcript level changes and function of TaNRT2s (Figs. 6, 7 and 8). Transcriptome analysis showed that the genes involved in nitrogen metabolism were observably changed under nitrate-limited conditions (Fig. 6) and approximately half of the wheat NRT2 genes were upregulated in roots under nitrate stress (Fig. 7). We selected two different NUE wheat varieties and identified the expression levels of three key TaNRT2 genes (TaNRT2-6A.2, TaNRT2-6A.6, TaNRT2-6B.4) in the two different NUE wheat varieties (Fig. 8). These three genes were highly expressed in nitrogen efficient material. Furthermore, we identified the function of the three genes in nitrate uptake which marked with 15N in Xenopus oocytes. The results showed that compared with water controls, single injection of TaNRT2-6B.4 observably increased in 15N accumulation, while TaNRT2-6A.2 and TaNRT2-6A.6 were indistinguishable with control (Fig. S3). It was reported that NRT2 proteins need form complexes with NRT3 to target the plasma membrane and maintain protein stability [38], and we believe there exist the same mechanism in wheat. In additional, several TaNRT2 genes (TaNRT2-7A/7B/7D) were observably induced in shoots, which suggests that these genes may function in nitrate distribution, and several TaNRT2 genes (TaNRT2-3A/3B/3D) were observably induced in seeds which suggests that these genes may participate in nitrogen accumulation in grains (Fig. 9). Interestingly, almost half of the TaNRT2 genes showed low expression or were not expressed in the roots and other tissues with or without nitrate deficiency treatment (Figs. 5 and 7). We speculated that these genes may be differentiated due to the functional redundancy of the large NRT2 gene family in wheat.

Model of expression patterns and induced TaNRT2 genes in wheat under nitrogen deficiency condition. The induced TaNRT2 genes were summarized in the model which based on the transcript data and the heatmap of TaNRT2 genes in different developmental stage. Under nitrate deficiency condition, most TaNRT2 genes are induced in root, these high-affinity nitrate transporters uptake NO3− by trans-membrane. Then the nitrate were distributed in leaves and seeds to assimilation. The red arrows indicate nitrate uptake and transport. The green ellipse on the membrane characterizes TaNRT2 proteins and the little yellow ellipses response nitrate molecules
NODULE INCEPTION (NIN) is functionally necessary for nodule formation in the legume Lotus japonicas, and its homologous gene is known as NINLIKE PROTEIN (NLP) [39]. Recent studies have highlighted the emerging roles of NLPs in N signalling and assimilation, root cap release [40,41,42], and regulation of nitrate uptake/transport under low- and high-nitrate conditions by combining to NRT2s [43, 44]. In this study, we analysed the gene expression profiles of 18 TaNLP genes (Fig. S4). Interestingly, we found that several TaNLP genes were highly expressed in seeds and roots. To further analyse the relationship between TaNRT2s and TaNLPs, we cloned several TaNRT2s and TaNLPs which highly expressed in roots (TaNRT2-6A.6, TaNRT2-3D, TaNLP7) and seeds (TaNRT2-3D, TaNLP1, TaNLP3, TaNLP4) for yeast one-hybrid assay. The results showed that they can interact, suggesting that TaNLPs may interact with TaNRT2s to maintain nitrogen homeostasis in wheat (Fig. S5). The finding may provide a basis for future studies concerning the roles of the TaNRT2s and TaNLPs in wheat.
Post-translational modification (PTM) plays a key role in cellular biological functions and it has been reported that protein phosphorylation represents 53.5% of all PTMs [45, 46]. The high-affinity transporter AtNRT2.1 has been shown to be rapidly de-phosphorylated after 3 min of nitrate resupply [47]. Subsequent studies have revealed that substitution of Ser28 resulted in unstable and de-phosphorylated AtNRT2.1 that failed to complement the growth‐restricted phenotype of the nrt2.1 mutant under low nitrate supply [48]. Another key phospho-site for AtNRT2.1 activity is Ser501, which can inactivate AtNRT2.1 function when mimicking the constitutive phosphorylation of this residue in transgenic plants [49]. Based on these results, we hypothesized that the two phosphor-sites may be highly conserved. We analysed all the NRT2 proteins by amino acid sequence alignment in wheat, rice, maize, and Arabidopsis (Additional file 1). The results showed that phosphor-site Ser28 was not conserved in rice, maize, and wheat, but was conserved in Arabidopsis. Another phospho-site, Ser501, was very conserved in maize and rice, except for OsNRT2.4, whereas it was only partly conserved in wheat. It has been reported that a large-scale expansion of NRT2 genes has occurred in Triticeae and is mainly concentrated on chromosome 6 [37]. In this study, 35 TaNRT2 genes located on chromosome 6 and three TaNRT2 genes located on chromosome 7 were not conserved for Ser501, while other TaNRT2 genes located on chromosomes 1, 2 and 3 were conserved for Ser501. We suggest that those that did not contain a conserved Ser501 site on chromosome 6 may have originated from a single gene before large-scale expansion, and that this original gene did not contain the conserved site.
N and phosphorus (P) are the two most important mineral nutrients for plants. It has been reported that variations in the N:P supply ratio significantly affect their uptake and an increased N:P supply ratio greatly promotes the uptake of P [50, 51]. PHOSPHORUS STARVATION RESPONSE 1 (PHR1) is a key transcription factor involved in phosphate starvation signalling [52]. In recent years, many studies have demonstrated that PHR1 also plays an important role in nitrogen nutrition. In rice, PHR2-SPX4 and NLP3 activate both phosphate- and nitrate-responsive genes. This leads to the coordinated utilization of nitrogen and phosphorus [53]. In Arabidopsis, PHR1 and NIGT1 together regulate the acquisition of phosphorus and nitrogen [54]. In this study, the transcriptome data showed that a few TaPHR1s and other phosphate related genes notably changed under nitrate deficiency (Table S1). This implies that these genes play important roles in the response to phosphorus and nitrogen balance. However, verification of this finding requires further research.
Conclusions
In summary, 49 TaNRT2 genes distributed on 13 chromosomes were identified in the wheat genome. A hypothetical model of all the TaNRT2s involved in nitrate absorption, distribution, and accumulation is proposed and is based on the transcriptome analysis and the expression profiles of wheat throughout its growth and development stages (Fig. 9). In particular, several genes were specifically expressed in the roots, leaves and seeds and strongly induced by nitrogen deficiency stress. This analysis of the function of these genes will improve the NUE in wheat. However, further research is needed to clarify the nitrogen assimilation mechanism in wheat.
Methods
Plant growth conditions and low NO3- stress treatment
Three wheat varieties, ‘Chuanmai104’, ‘Nanmai660’ and ‘Mianmai367’ which are the cultivars of Southwest China were used in this study. They were cultured hydroponically in a growth chamber under the following conditions: relative humidity, 50–70%; 14-h light/10-h dark photoperiod; temperatures, 22 °C days, 22 °C nights.
A modified Hoagland nutrient solution was used in this study [42], with 5 mM KNO3 as sufficient nitrogen (HN) solution and 0.1 mM KNO3 or 0 mM KNO3 as LN solution. The solutions were changed every second day. The solution for HN conditions contained 5 mM KNO3, 1 mM KH2PO4, 2 mM MgSO4, 0.1 mM FeNaEDTA, 5 μM KI, 1 μM H3BO3, 0.15 mM MnSO4, 0.05 mM ZnSO4, 4 mM CaCl2, 0.19 mM CoCl2, 0.1 μM CuSO4 and 1 μM Na2MoO4, (pH = 5.8). The solution for the LN condition contained the same nutrients with the removal of KNO3, and the differences in potassium supply were balanced with KCl.
Identification of the NRT2 gene family in wheat
To identify putative NRT2 genes in wheat, the known wheat NRT2 protein sequence (AAG01172.1), downloaded from NCBI (https://www.ncbi.nlm.nih.gov/), was queried by blastp on WheatOmics 1.0 [55] using the IWGSC RefSeq v1.1 (Chinese Spring) genome database (Additional file 2). The Pfam online server (http://pfam.xfam.org/search) was used to predict the conserved domains, and the TMHMM-2.0 online server (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0) was used to predict NRT2 protein transmembrane helices. The sequences were processed by removing the non-conserved MFS_1 domain and less than 6 transmembrane helices, and after manual curation, a final set of 49 genes belonging to the NTR2 nitrate transporter family were selected. The NTR2 protein feature prediction, molecular weight and theoretical protein isoelectric point (pI) were predicted by ProtParam (https://web.expasy.org/protparam/), and the subcellular localization prediction was analysed by POST (http://psort1.hgc.jp/form.html). The chromosome location of TaNRT2 genes were visualized by TBtools [56] based on the IWGSC RefSeq v1.1 wheat genome database. The collinearity was determined by MCScanX toolkit [57] using the wheat genomic DNA sequence and the gff3 file.
Phylogenetic analysis of TaNRT2
The protein sequences of A. thaliana, O. sativa and Z. mays were obtained from a reported study [10, 16,17,18], and these protein loci are listed in Additional file 3. The full-length proteins of AtNRT2s, OsNRT2s, ZmNRT2s, and the newly identified TaNRT2s were aligned using ClustalW, and the phylogenetic tree was constructed based on the alignment using MEGA7 [58] by using neighbor-joining (NJ) algorithms with the following parameters: Jones-Taylor-Thornton (JTT) model, pairwise deletion and bootstrap (1,000 replicates), and visualization by Evolview 2.0 [59].
Analysis of motifs, domain and gene structure
Protein motifs were identified by using MEME (Multiple Expectation Maximization for Motif Elication) (https://meme-suite.org/meme/tools/meme). Conserved domains were identified by NCBI CDD (https://www.ncbi.nlm.nih.gov/cdd/?term=). Gene structure information was extracted from the gff3 file for the wheat reference genome (IWGSC RefSeq v1.1). The characteristics of the TaNRT2 family gene structure with motif composition and conserved domains were visualized by TBtools.
Cis-acting regulatory element (CARE) analysis
Cis-acting regulatory elements (CAREs) were predicted by using the 2000 bp upstream region of wheat NRT2 genes in a Plant CARE online server (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) and the distribution of CAREs on the gene promoter was visualized by TBtools.
Expression profiling of all TaNRT2 and TaNLP genes
The TPM values of wheat NRT2 and NLP genes come from five tissues (root, stem, leaf, spike and grain) were obtained from the Wheat Expression Browser on the WheatOmics 1.0 online server [55]. All the TPM values were logarithmic and visualized as heatmaps of TaNRT2s and TaNLPs using the TBtools integrated toolkit. The FPKM values of all TaNRT2 genes from transcriptome databases after nitrogen deficiency treatment were used to construct the heatmap using the TBtools integrated toolkit.
Biomass and nitrogen content measurement
The two wheat seedlings of LN (0.1 mM NO3−) and HN (5 mM NO3−) treated in hydroponics were collected after 12 days. The nutrient solution was replaced every second day. The root and shoot tissues were harvested separately and dried at 80 °C for 3 days, and then the dry weights were recorded. The dried samples were powdered and subsequently digested with concentrated H2SO4 for the determination of total N using the Kjeldahl method [60].Three biological replicates were used for phenotypic tests, biomass and nitrogen content measurements. The t test (* P < 0.05, ** P < 0.01) was used to analyse the statistical significance.
RNA-seq analysis
The seeds of ‘Chuanmai104’ were germinated and grown on vermiculite for 15 days to two-leaf stage, then seedlings were transferred to modified Hoagland hydroponic solution grown for 3 days. First, the wheat seedlings were cultured in normal solution for 3 days. Then, half of them were transferred to nitrogen starvation conditions (0 mM nitrate) as the NO3−-deficient treatment (LN), and the other half were transferred to normal solution (5 mM nitrate) as the control (HN) in hydroponics. After 3 days, the wheat seedlings of shoots and roots were collected, and three biological replicates were used for RNA-seq analysis (Novogene, China). Differential expression analysis of two conditions was performed using the DESeq2R package (1.20.0). DESeq2 provides statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting P values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate. Genes with an adjusted P value ≦0.05 found by DESeq2 were defined as differentially expressed genes. The raw date of transcriptome data are shown in Additional files 4 and 5.
Gene Ontology (GO) enrichment analysis of differentially expressed genes was implemented by the cluster Profiler R package, in which gene length bias was corrected. GO terms with corrected P values less than 0.05 were considered significantly enriched by differentially expressed genes. KEGG is a database resource for understanding the high-level functions and utilities of biological systems [61], such as the cells, the organisms and ecosystems, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies (www.kegg.jp/kegg/kegg1.html). We used the clusterProfiler R package to test the statistical enrichment of differentially expressed genes in KEGG pathways.
Uptake of K15NO3- in Xenopus oocytes
Coding sequences of TaNRT2-6A.2, TaNRT2-6A.6, and TaNRT2-6B.4 were cloned into the expression vector pT7TS. After linearization of pT7TS plasmids with EcoRI, RNA was transcribed in vitro using an mRNA synthesis kit (mMESSAGE mMACHINE T7 kit; Ambion). Xenopus laevis oocytes were injected with 25 ng RNA and incubated for 60 h in ND96 solution (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES, pH 7.5).
For 15N uptake in oocytes, ten oocytes were selected treatment in NO3− uptake solution (230 mM mannitol, 0.3 mM CaCl2, 10 mM MES with 0.25 mM K15NO3−) for 12 h at 18 °C and washed five times in ND96 solution. 15N was measured using an isotope ratio mass spectrometer (IRMS; DELTAplus XP) according to previous report [38, 62].
Yeast one-hybrid assay
The coding sequence of TaNLPs was constructed into the vector pB42AD. LacZ was used as a reporter gene, driven by the fragments of TaNRT2s promoter in yeast. The pB42AD-TaNLPs and PB42AD plasmids were transformed with the pTaNRT2s:lacZ plasmids into Saccharomyces cerevisiae strain EGY48 using the PEG/LiAC method. The transformed strains were cultured on SD/-Trp-Ura plates and confirmed by PCR. Then, these transformants were grown on proper SD/-Trp-Ura plates containing X-α-gal (5-bromo-4-chloro-3-indolyl-α-D-galactopyranoside), 2% galactose, and 1% raffinose for blue colour development [63].
RNA isolation and real-time PCR analysis
The shoots and roots of wheat seedlings were treated with nitrate limited conditions (0.1 mM nitrate) and complete nutrient solution (5 mM nitrate) for 12 days, and these seedlings were collected and immediately frozen in liquid nitrogen and stored at − 80 °C. The total RNA of wheat seedlings was extracted with RNA extraction kit (EASYspin Plus Complex Plant RNA Kit) [64], and treated with DNase I (Takara, Dalian, China) to eliminate genomic DNA contamination. Then, the total RNA was used to synthesize cDNA with a reverse transcription reaction kit (Thermo Scientific, Lithuania). The qRT‒PCR assay was conducted as described previously [65]. Amplification of wheat α-tubulin gene was used as an internal control to normalize the data. The primers used are listed in Table S2. The gene-specific primers were designed using NCBI (Primer designing tool (nih.gov)) and DNAMAN software.
Availability of data and materials
All data generated or analyzed during this study are included in this article and additional files. The transcriptome sequence were generated in Novogene company (https://cn.novogene.com/). The datasets generated during the current study are available in the National Center for Biotechnology Information Sequence Read Archive (SRA) under accession number PRJNA925925 (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA925925&o=acc_s%3Aa). The datasets supporting the conclusions of this article are included within the article (and its additional files).
Abbreviations
- DEG:
-
Differently expressed gene
- RT-PCR:
-
Real-time polymerase chain reaction
- NUE:
-
Nitrogen utilization efficiency
- NRT:
-
Nitrate transporter
- MFS:
-
The major facilitator superfamily
- LN:
-
Low nitrogen
- HN:
-
High nitrogen
- LATSs:
-
Low-affinity nitrate transporter systems
- HATSs:
-
High-affinity nitrate transporter systems
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- FPKM:
-
Fragments per kilobase of exon per million fragments mapped
- TPM:
-
Transcripts per million
- PTM:
-
Post-translational modification
References
Wang YY, Hsu PK, Tsay YF. Uptake, allocation and signaling of nitrate. Trends Plant Sci. 2012;17(8):458–67.
Kant S. Understanding nitrate uptake, signaling and remobilisation for improving plant nitrogen use efficiency. Semin Cell Dev Biol. 2018;74:89–96.
Fradgley NS, Bentley AR, Swarbreck SM. Defining the physiological determinants of low nitrogen requirement in wheat. Biochem Soc Trans. 2021;49(2):609–16.
Liu C, Chen F, Li Z, Cocq KL, Liu Y, Wu L. Impacts of nitrogen practices on yield, grain quality, and nitrogen-use efficiency of crops and soil fertility in three paddy-upland cropping systems. J Sci Food Agric. 2021;101(6):2218–26.
Hawkesford MJ. Genetic variation in traits for nitrogen use efficiency in wheat. J Exp Bot. 2017;68(10):2627–32.
Good AG, Beatty PH. Fertilizing nature: a tragedy of excess in the commons. PLoS Biol. 2011;9(8):e1001124.
Zhang J, Zhang H, Li S, Li J, Yan L, Xia L. Increasing yield potential through manipulating of an ARE1 ortholog related to nitrogen use efficiency in wheat by CRISPR/Cas9. J Integr Plant Biol. 2021;63(9):1649–63.
Shcherbak I, Millar N, Robertson GP. Global metaanalysis of the nonlinear response of soil nitrous oxide (N2O) emissions to fertilizer nitrogen. Proc Natl Acad Sci U S A. 2014;111(25):9199–204.
Xu G, Fan X, Miller AJ. Plant nitrogen assimilation and use efficiency. Annual Rev Plant Biol. 2012;63:153–82.
Wang YY, Cheng YH, Chen KE, Tsay YF. Nitrate transport, signaling, and use efficiency. Annual Rev Plant Biol. 2018;69:85–122.
Fan X, Naz M, Fan X, Xuan W, Miller AJ, Xu G. Plant nitrate transporters: from gene function to application. J Exp Bot. 2017;68(10):2463–75.
Crawford NM, Glass ADM. Molecular and physiological aspects of nitrate uptake in plants. Trends Plant Sci. 1998;3(10):389–95.
Vidal EA, Alvarez JM, Araus V, Riveras E, Brooks MD, Krouk G, Ruffel S, Lejay L, Crawford NM, Coruzzi GM, et al. Nitrate in 2020: Thirty years from transport to signaling networks. Plant Cell. 2020;32(7):2094–119.
Brownlee AG, Arst HN Jr. Nitrate uptake in Aspergillus nidulans and involvement of the third gene of the nitrate assimilation gene cluster. Trends Plant Sci. 1983;155(3):1138–46.
Unkles SE, Hawker KL, Grieve C, Campbell EI, Montague P, Kinghorn JR. crnA encodes a nitrate transporter in Aspergillus nidulans. Proc Natl Acad Sci U S A. 1991;88(1):204–8.
Orsel M, Krapp A, Daniel-Vedele F. Analysis of the NRT2 nitrate transporter family in Arabidopsis. Structure and gene expression. Plant Physiol. 2002;129(2):886–96.
Cai C, Wang JY, Zhu YG, Shen QR, Li B, Tong YP, Li ZS. Gene structure and expression of the high-affinity nitrate transport system in rice roots. J Integr Plant Biol. 2008;50(4):443–51.
Plett D, Toubia J, Garnett T, Tester M, Kaiser BN, Baumann U. Dichotomy in the NRT gene families of dicots and grass species. PLoS one. 2010;5(12):e15289.
Du RJ, Wu ZX, Yu ZX, Li PF, Mu JY, Zhou J, Li JN, Du H. Genome-wide characterization of high-affinity nitrate transporter 2 (NRT2) gene family in Brassica napus. Int J Mol Sci. 2022;23(9):4965.
Tong J, Walk TC, Han P, Chen L, Shen X, Li Y, Gu C, Xie L, Hu X, Liao X, et al. Genome-wide identification and analysis of high-affinity nitrate transporter 2 (NRT2) family genes in rapeseed (Brassica napus L.) and their responses to various stresses. BMC Plant Biol. 2020;20(1):464.
Zhao L, Chen P, Liu P, Song Y, Zhang D. Genetic effects and expression patterns of the nitrate transporter (NRT) gene family in Populus tomentosa. Front Plant Sci. 2021;12:661635.
Akbudak MA, Filiz E, Cetin D. Genome-wide identification and characterization of high-affinity nitrate transporter 2 (NRT2) gene family in tomato (Solanum lycopersicum) and their transcriptional responses to drought and salinity stresses. J Plant Physiol. 2022;272:153684.
You H, Liu Y, Minh TN, Lu H, Zhang P, Li W, Xiao J, Ding X, Li Q. Genome-wide identification and expression analyses of nitrate transporter family genes in wild soybean (Glycine soja). J Appl Genet. 2020;61(4):489–501.
Cerezo M, Tillard P, Filleur S, Muños S, Daniel-Vedele F, Gojon A. Major alterations of the regulation of root NO3- uptake are associated with the mutation of Nrt2.1 and Nrt2.2 genes in Arabidopsis. Plant Physiol. 2001;127(1):262–71.
Li W, Wang Y, Okamoto M, Crawford NM, Siddiqi MY, Glass AD. Dissection of the AtNRT2.1:AtNRT2.2 inducible high-affinity nitrate transporter gene cluster. Plant Physiol. 2007;143(1):425–33.
Kiba T, Feria-Bourrellier A-B, Lafouge F, Lezhneva L, Boutet-Mercey S, Orsel M, Bréhaut V, Miller A, Daniel-Vedele F, Sakakibara H, et al. The Arabidopsis nitrate transporter NRT2.4 plays a double role in roots and shoots of nitrogen-starved plants. Plant Cell. 2012;24(1):245–58.
Lezhneva L, Kiba T, Feria-Bourrellier AB, Lafouge F, Boutet-Mercey S, Zoufan P, Sakakibara H, Daniel-Vedele F, Krapp A. The Arabidopsis nitrate transporter NRT2.5 plays a role in nitrate acquisition and remobilization in nitrogen-starved plants. Plant J. 2014;80(2):230–41.
Chopin F, Orsel M, Dorbe MF, Chardon F, Truong HN, Miller AJ, Krapp A, Daniel-Vedele F. The Arabidopsis ATNRT2.7 nitrate transporter controls nitrate content in seeds. Plant Cell. 2007;19(5):1590–602.
Feng H, Yan M, Fan X, Li B, Shen Q, Miller AJ, Xu G. Spatial expression and regulation of rice high-affinity nitrate transporters by nitrogen and carbon status. J Exp Bot. 2011;62(7):2319–32.
Tang Z, Fan X, Li Q, Feng H, Miller AJ, Shen Q, Xu G. Knockdown of a rice stelar nitrate transporter alters long-distance translocation but not root influx. Plant Physiol. 2012;160(4):2052–63.
Fan X, Tang Z, Tan Y, Zhang Y, Luo B, Yang M, Lian X, Shen Q, Miller AJ, Xu G. Overexpression of a pH-sensitive nitrate transporter in rice increases crop yields. Proc Natl Acad Sci U S A. 2016;113(26):7118–23.
Wei J, Zheng Y, Feng H, Qu H, Fan X, Yamaji N, Ma JF, Xu G. OsNRT2.4 encodes a dual-affinity nitrate transporter and functions in nitrate-regulated root growth and nitrate distribution in rice. J Exp Bot. 2018;69(5):1095–107.
Li W, He X, Chen Y, Jing Y, Shen C, Yang J, Teng W, Zhao X, Hu W, Hu M, et al. A wheat transcription factor positively sets seed vigour by regulating the grain nitrate signal. New Phytol. 2020;225(4):1667–80.
Lupini A, Mercati F, Araniti F, Miller AJ, Sunseri F. Abenavoli MR NAR2.1/NRT2.1 functional interaction with NO3- and H+ fluxes in high-affinity nitrate transport in maize root regions. Plant Physiol Biochem. 2016;102:107–14.
Higo K, Ugawa Y, Iwamoto M, Korenaga T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999;27(1):297–300.
Kumar A, Sandhu N, Kumar P, Pruthi G, Singh J, Kaur S, Chhuneja P. Genome-wide identification and in silico analysis of NPF, NRT2, CLC and SLAC1/SLAH nitrate transporters in hexaploid wheat (Triticum aestivum). Sci Rep. 2022;12(1):11227.
Shi X, Cui F, Han X, He Y, Zhao L, Zhang N, Zhang H, Zhu H, Liu Z, Ma B, et al. Comparative genomic and transcriptomic analyses uncover the molecular basis of high nitrogen-use efficiency in the wheat cultivar Kenong 9204. Mol Plant. 2022;15(9):1440–56.
Kotur Z, Mackenzie N, Ramesh S, Tyerman SD, Kaiser BN, Glass ADM. Nitrate transport capacity of the Arabidopsis thaliana NRT2 family members and their interactions with AtNAR2.1. New Phytol. 2012;194(3):724–31.
Chardin C, Girin T, Roudier F, Meyer C, Krapp A. The plant RWP-RK transcription factors: key regulators of nitrogen responses and of gametophyte development. J Exp Bot. 2014;65(19):5577–87.
Yan D, Easwaran V, Chau V, Okamoto M, Ierullo M, Kimura M, Endo A, Yano R, Pasha A, Gong Y, et al. NIN-like protein 8 is a master regulator of nitrate-promoted seed germination in Arabidopsis. Nat Commun. 2016;7:13179.
Alfatih A, Wu J, Zhang ZS, Xia JQ, Jan SU, Yu LH, Xiang CB. Rice NIN-LIKE PROTEIN 1 rapidly responds to nitrogen deficiency and improves yield and nitrogen use efficiency. J Exp Bot. 2020;71(19):6032–42.
Ge M, Wang Y, Liu Y, Jiang L, He B, Ning L, Du H, Lv Y, Zhou L, Lin F, et al. The NIN-like protein 5 (ZmNLP5) transcription factor is involved in modulating the nitrogen response in maize. Plant J. 2020;102(2):353–68.
Luo Z, Wang J, Li F, Lu Y, Fang Z, Fu M, Mysore KS, Wen J, Gong J, Murray JD, et al. The small peptide CEP1 and the NIN-like protein NLP1 regulate NRT2.1 to mediate root nodule formation across nitrate concentrations. Plant Cell. 2023;35(2):776–94.
Misawa F, Ito M, Nosaki S, Nishida H, Watanabe M, Suzuki T, Miura K, Kawaguchi M, Suzaki T. Nitrate transport via NRT2.1 mediates NIN-LIKE PROTEIN-dependent suppression of root nodulation in Lotus japonicus. Plant Cell. 2022;34(5):1844–62.
Silva-Sanchez C, Li H, Chen S. Recent advances and challenges in plant phosphoproteomics. Proteomics. 2015;15(5–6):1127–41.
Wang W, Li A, Zhang Z, Chu C. Posttranslational modifications: regulation of nitrogen utilization and signaling. Plant Cell Physiol. 2021;62(4):543–52.
Engelsberger WR, Schulze WX. Nitrate and ammonium lead to distinct global dynamic phosphorylation patterns when resupplied to nitrogen-starved Arabidopsis seedlings. Plant J. 2012;69(6):978–95.
Zou X, Liu MY, Wu WH, Wang Y. Phosphorylation at Ser28 stabilizes the Arabidopsis nitrate transporter NRT2.1 in response to nitrate limitation. J Integr Plant Biol. 2020;62(6):865–76.
Jacquot A, Chaput V, Mauries A, Li Z, Tillard P, Fizames C, Bonillo P, Bellegarde F, Laugier E, Santoni V, et al. NRT2.1 C-terminus phosphorylation prevents root high affinity nitrate uptake activity in Arabidopsis thaliana. New Phytol. 2020;228(3):1038–54.
Gusewell S. N : P ratios in terrestrial plants: variation and functional significance. New Phytol. 2004;164(2):243–66.
Luo X, Mazer SJ, Guo H, Zhang N, Weiner J, Hu S. Nitrogen:phosphorous supply ratio and allometry in five alpine plant species. Ecol Evol. 2016;6(24):8881–92.
Rubio V, Linhares F, Solano R, Martin AC, Iglesias J, Leyva A, Paz-Ares J. A conserved MYB transcription factor involved in phosphate starvation signaling both in vascular plants and in unicellular algae. Genes Dev. 2001;15(16):2122–33.
Hu B, Jiang Z, Wang W, Qiu Y, Zhang Z, Liu Y, Li A, Gao X, Liu L, Qian Y, et al. Nitrate-NRT1.1B-SPX4 cascade integrates nitrogen and phosphorus signalling networks in plants. Nat Plants. 2019;5(4):401–13.
Maeda Y, Konishi M, Kiba T, Sakuraba Y, Sawaki N, Kurai T, Ueda Y, Sakakibara H, Yanagisawa S. A NIGT1-centred transcriptional cascade regulates nitrate signalling and incorporates phosphorus starvation signals in Arabidopsis. Nat Commun. 2018;9(1):1379.
Ma S, Wang M, Wu J, Guo W, Chen Y, Li G, Wang Y, Shi W, Xia G, Fu D, et al. WheatOmics: a platform combining multiple omics data to accelerate functional genomics studies in wheat. Mol Plant. 2021;14(12):1965–8.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49.
Kumar S, Stecher G, Tamura K. MEGA7:molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
He Z, Zhang H, Gao S, Lercher MJ, Chen WH, Hu S. Evolview v2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016;44(W1):W236-241.
Thomas RL, Sheard RW, Moyer JR. Comparison of conventional and automated procedures for nitrogen, phosphorus, and potassium analysis of plant material using a single digestion. Agron J. 1967;59(3):240–3.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587-d592.
Li H, Yu M, Du XQ, Wang ZF, Wu WH, Quintero FJ, Jin XH, Li HD, Wang Y. NRT1.5/NPF7.3 functions as a proton-coupled H+/K+ antiporter for K+ loading into the xylem in Arabidopsis. Plant Cell. 2017;29(8):2016–26.
Wang ZF, Mi TW, Gao YQ, Feng HQ, Wu WH, Wang Y. STOP1 regulates LKS1 transcription and coordinates K+/NH4+ balance in Arabidopsis response to low-K+ stress. Int J Mol Sci. 2021;23(1):383.
Wang F, Tan WF, Song W, Yang ST, Qiao S. Transcriptome analysis of sweet potato responses to potassium deficiency. BMC Genomics. 2022;23(1):655.
Wang F, Cui PJ, Tian Y, Huang Y, Wang HF, Liu F, Chen YF. Maize ZmPT7 regulates Pi uptake and redistribution which is modulated by phosphorylation. Plant Biotechnol J. 2020;18(12):2406–19.
Acknowledgements
We thank Nature Research Editing Service (https://authorservices.springernature.com/language-editing/) for the English language polishing. The authors thank professor Yang Wang from the China Agricultural University for providing the 15N uptake assay technology in Xenopus oocytes.
Funding
This work was financially supported by Sichuan Wheat Breeding Community (2021YFYZ0002-02) to perform research design and most of the experiments, the 1 + 9 open Competition Project of Sichuan Academy of Agricultural Sciences (1 + 9KJGG002) funded us in bioinformatics and data analysis, Sichuan Science and Technology Program (2022JDRC0113) and Sichuan Provincial Finance Project (2021ZYGG-003) had no influence over the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
Fang Wang and Qing-Yan Deng conducted the experiments, analyzed the results and performed the figures. Fang Wang, Zong-Jun Pu and Wen-Fang Tan designed and wrote the paper. Jiang-Tao Luo and Jian-Min Zheng assisted in the experimental procedures. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The wheat varieties used in this study (‘Chuanmai104’, ‘Mianmai367’, ‘Nanmai660’) were both acquired from Crop Research Institute, Sichuan Academy of Agricultural Sciences, Chengdu, China. All the wheat varieties in this study were permitted and have no conflict of interest. All methods were performed in accordance with the relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
A multiple sequence alignment of all the TaNRT2 proteins.
Additional file 2.
The NRT2 genes information in wheat.
Additional file 3.
The amino acid sequence of NRT2s in Arabidopsis, maize, rice and wheat.
Additional file 4.
The raw data of transcriptome in root under nitrogen deficiency condition.
Additional file 5.
The raw data of transcriptome in shoot under nitrogen deficiency condition.
Additional file 6: Fig. S1.
The number and ratio of NRT2 genes in wheat, rice, maize and Arabidopsis. a The number of NRT2 genes in wheat genome and sub-genome, rice, maize and Arabidopsis. b The ratio of total NRT2 gene is shown for wheat : rice (red) and wheat : Arabidopsis (orange). The expected ratio (3 : 1) is indicated by a black dotted line. Fig. S2. Gene classification was based on GO analysis for DEGs under nitrate deficiency condition. The numbers of DEGs in each GO term was significantly enriched in root (a) and shoot (b). Functional categorization of genes based on the biological process of gene ontology. Different classes are shown for BP (biological process) ,CC (cellular component) and MF (molecular function). The y‑axis shows the counts of differently expressed genes, and the x‑axis shows GO term of gene enriched in each biological process. Fig. S3. K15NO3 uptake into Xenopus oocytes. oocytes injected with water as control, cRNA of TaNRT2-6A.2, TaNRT2-6A.6, TaNRT2-6B.4 were injected alone, respectively.15N enrichment per oocyte is expressed as delta 15N compared with standard atmospheric 15N : 14N ratio. Values are average of n = 6 ± SD. Differences between mean values of treatments and controls were compared using t - tests (* P< 0.05). Fig. S4. Heatmap representing the expression pattern of TaNLP genes in various developmental stages. The TPM values normalized by logarithmic scale were used to construct the heatmap. Z10~Z85 represent different growth stage of wheat. Different colors represent relative expression levels, as shown in the legend on the right. The horizontal axis represents the names and classifications genes, and the vertical axis represents various tissues. The rows of the heat map are clustered according to the expression patterns. Fig. S5. Yeast one-hybrid (Y1H) assay was used to verify TaNLPs bound to the TaNRT2s promoter region. TaNLPs fusion proteins activate the expression of LacZ reporter gene driven by the promoter of TaNRT2-6A.6 and TaNRT2-3D, respectively, in yeast. The empty vector pB42AD was used as a negative control. Table S1. Phosphorus signaling pathway genes respond to low nitrogen stress. Table S2. Primers used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Deng, QY., Luo, JT., Zheng, JM. et al. Genome-wide systematic characterization of the NRT2 gene family and its expression profile in wheat (Triticum aestivum L.) during plant growth and in response to nitrate deficiency. BMC Plant Biol 23, 353 (2023). https://doi.org/10.1186/s12870-023-04333-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04333-5