
Abstract
Background
Drought stress limits the production of soybean [Glycine max (L.) Merr.], which is the most grown high-value legume crop worldwide. Breeding for drought tolerance is a difficult endeavor and understanding the genetic basis of drought tolerance in soybean is therefore crucial for harnessing the genomic regions involved in the tolerance mechanisms. A genome-wide association study (GWAS) analysis was applied in a soybean germplasm collection (the EUCLEG collection) of 359 accessions relevant for breeding in Europe, to identify genomic regions and candidate genes involved in the response to short duration and long duration drought stress (SDS and LDS respectively) in soybean.
Results
The phenotypic response to drought was stronger in the long duration drought (LDS) than in the short duration drought (SDS) experiment. Over the four traits considered (canopy wilting, leaf senescence, maximum absolute growth rate and maximum plant height) the variation was in the range of 8.4−25.2% in the SDS, and 14.7−29.7% in the LDS experiments. The GWAS analysis identified a total of 17 and 22 significant marker-trait associations for four traits in the SDS and LDS experiments, respectively. In the genomic regions delimited by these markers we identified a total of 12 and 16 genes with putative functions that are of particular relevance for drought stress responses including stomatal movement, root formation, photosynthesis, ABA signaling, cellular protection and cellular repair mechanisms. Some of these genomic regions co-localized with previously known QTLs for drought tolerance traits including water use efficiency, chlorophyll content and photosynthesis.
Conclusion
Our results indicate that the mechanism of slow wilting in the SDS might be associated with the characteristics of the root system, whereas in the LDS, slow wilting could be due to low stomatal conductance and transpiration rates enabling a high WUE. Drought-induced leaf senescence was found to be associated to ABA and ROS responses. The QTLs related to WUE contributed to growth rate and canopy height maintenance under drought stress. Co-localization of several previously known QTLs for multiple agronomic traits with the SNPs identified in this study, highlights the importance of the identified genomic regions for the improvement of agronomic performance in addition to drought tolerance in the EUCLEG collection.
Similar content being viewed by others


Introduction
Human population growth and climate change are major challenges for the world’s future food security [1, 2]. Incidence of extreme weather events, such as erratic rainfall, raising temperatures and the consequent higher chance of drought spells cause significant reductions in crop production [3]. This, in combination with the need to feed a growing population without increasing the environmental burden on natural ecosystems, requires innovative solutions in agriculture, including the selection of crops with improved tolerance to water deficit through breeding [4]. Soybean [Glycine max (L.) Merr.] is the most grown-high value legume crop, and delivers a large part of the increasing demand for protein and oil in the world [5]. Drought stress has been identified as one of the challenges limiting soybean production in different parts of the world [6,7,8,9]. Therefore, breeding soybean for drought tolerance is imperative to avoid yield losses [10].
Drought stress causes several physiological, biochemical and molecular changes in plants such as reduced photosynthesis [11], oxidative stress caused by the accumulation of reactive oxygen species (ROS) [12], alterations in the metabolism of enzymes and other cellular compounds [13] and transcriptional modifications [14]. These changes are reflected in traits linked to crop performance including reduced stem growth and plant height, leaf wilting and senescence, altered root growth and reduced yield [15,16,17]. Our knowledge of drought tolerance in crops is fragmentary, but it is known that multiple plant characteristics and genomic loci influence the ability of plants to withstand drought stress [10, 18,19,20]. Furthermore, the timing, duration and intensity of the drought stress situation can strongly influence the plant responses [21,22,23,24]. Therefore, understanding the genetic basis of drought tolerance is crucial to get a better insight in tolerance mechanisms and for harnessing the genomic regions involved for crop improvement [25,26,27].
The genetic control of drought tolerance has been studied in major crops and, in some cases, this knowledge has been implemented in breeding programs. For example, in wheat, genes behind the synthesis and regulation of different types of enzymes and compounds such as Late Embryogenesis Abundant (LEA) responsive to abscisic acid (Rab), RuBisCO, helicases, proline, glutathione-S-transferase (GST) and carbohydrates are involved in drought tolerance [28]. QTLs have also been identified for water use efficiency (WUE) assessed through the carbon isotope ratio (δ13C), concentration of water-soluble carbohydrates (WSC), root system properties, and grain yield recorded under water stress [18]. Furthermore, [29] reported the introgression of multiple QTLs related to canopy temperature, chlorophyll content, stay green habit, Normalized Difference Vegetation Index (NDVI) values, days to anthesis and grain yield through marker assisted backcross breeding in wheat. Studies in maize have mainly focused on identifying the genetic basis of seedling survival rate, interval from anthesis to silking and yield-related traits [30]. Maize genotypes introgressed with QTLs for a short anthesis to silking interval outperformed for yield under drought but the yield advantage decreased from severe to mild stress. This illustrates that drought may activate different molecular mechanisms depending on the intensity of the stress situation [31]. Drought tolerance in rice has been assessed through screening for leaf rolling, spikelet fertility, rooting depth, leaf relative water content and osmotic adjustment [32], and a number of QTLs underlying some of these traits have been used for genomics-assisted breeding. For example, two drought tolerance QTLs (qDTY2.2 and qDTY4.1) have been introgressed into the popular, high-yielding rice variety IR64 [33], and three QTLs (qDTY1.1, qDTY2.2 and qDTY4.1) have been introgressed into the Indian elite variety Nareen [34]. These introgressions confer yield advantage under drought, and no penalty under non-stress conditions [33, 34].
The genetic control of drought tolerance has also been studied in soybean. Canopy witling is one of the most often investigated traits in soybean in the context of adaptation to drought. Correspondingly, several QTLs have been reported along with candidate genes putatively involved in the regulation of transpiration and water conservation [35,36,37,38,39,40]. Also QTLs for canopy temperature [41, 42], plant height in drought relative to control treatments [43, 44] and yield-related traits [45,46,47] have been reported. Overall, it has been shown that drought tolerance in soybean is conferred by many QTLs, and most of the QTLs explain only a little part of the phenotypic variation [20].
Previous studies of drought tolerance in soybean mainly focused on the analysis of late maturing soybean genotypes (MG0-IV or later). It is therefore unclear whether the same kind of adaptations and genomic regions are of relevance for exploitation in early maturing types (MG000-II). These early maturing types are however of particular importance for European agriculture [48], where there is a growing interest in the cultivation of soybean, as one of the approaches to increase protein self-sufficiency [49]. Considering that the local soybean cultivation accounts for only 34% of the total 34.4 million tons consumed in Europe [50, 51], increasing the European soybean acreage can significantly contribute to reduce protein imports. This requires breeding soybean varieties well-adapted to the European environmental and cultivation conditions. While North West Europe is often considered a humid region, simulations have demonstrated that water can be the main limiting factor in soybean production, because in general water availability is not well distributed along the crop cycle [52]. In addition, drought spells associated with low rainfall and high temperature are becoming more frequent in Europe during the summer [53,54,55], and forecasts on climatic change predict that the frequency of these events will increase in the future [56]. It is therefore important to consider adaptation to drought in European soybean breeding programs.
Drought spells occurring at the developmental stages following flowering are critical in soybean. At these stages soybean plants require sufficient water to achieve their yield potential [57], and investigations on drought tolerance in soybean focus on this [58,59,60]. In previous work [23] we investigated the response to drought at the reproductive stage of a collection of early maturing soybean accessions (MG000-II) of relevance for breeding in Europe (the EUCLEG collection). Responses and traits of relevance for adaptation to drought, and their interactions, were identified. Furthermore, we demonstrated a differential response of the accessions to short duration and long duration drought stress [23]. The EUCLEG collection displayed a wide range of phenotypic variation in drought response for maximum absolute growth rate (AGRmax), maximum canopy height (CH), leaf senescence (LSEN) and canopy wilting (CW). The long duration drought treatment (for 6–7 weeks) caused a much stronger response than the short duration drought (for 3–4 weeks). Main responses were an average reduction of 11–29% in CH, an average reduction of 22% in AGRmax and an acceleration of the rate of senescence by 26–110%. Drought stress accelerated the plant development. A better tolerance for seed yield was associated with an earlier cessation of flowering and pod formation under short duration drought stress, and with the maintenance of AGRmax and CH under long duration drought stress. Stronger signs of LSEN and CW helped also some accessions to protect their yield under long duration drought stress. This study provided a set of traits (AGRmax, CH, LSEN and CW) that might be used to improve drought tolerance of early maturing European soybean, what justifies a further exploration of the genetic control of these traits.
Genome-wide association (GWAS) is a widely used approach to study the genetic basis of phenotypic variation in crops [61], and several QTLs for important traits have been identified through GWAS in soybean [40,41,42, 62, 63]. A prerequisite for this is the availability of high-resolution genotypic data. As reported in [64], the EUCLEG collection has been genotyped using the NJAU 355K SoySNP array and a detailed genetic diversity analysis on the basis of 225K polymorphic SNPs (Single Nucleotide Polymorphisms) has been performed.
Here we build further on these two studies [23, 64] and report on a GWAS analysis on drought tolerance in the EUCLEG soybean collection. Our main goal is to understand the genetic control of drought tolerance in this collection of early maturing types. More specifically, we aim for the identification of (i) SNPs and QTLs related to drought response for canopy wilting, leaf senescence, maximum absolute growth rate and maximum plant height and (ii) the candidate genes underlying these QTLs and (iii) mechanisms of tolerance under short duration and long duration drought stress.
Results
The results are based on separate analyses for the data collected in 2018 and 2019 because of the different weather conditions for these two years, causing a differential response of the accessions (for further details see [23]). For simplicity, here we refer to the experiments of 2018 and 2019 as the short-duration drought stress experiment (SDS) and the long-duration drought stress experiment (LDS) respectively.
Variation in the drought response
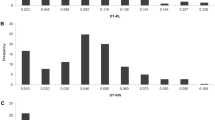
Basic statistics for the data of the four traits considered in this study (CW, LSEN, AGRmax-Yr and CH-Yr) are summarized in Table 1 and Figure 1. In general, the response to drought was stronger in the long-duration stress experiment (LDS), as indicated by higher average and maximum values recorded for the traits investigated, except for AGRmax-Yr (Table 1). Also the coefficient of variation was higher for all traits in the LDS experiment, except for CH-Yr (CV 24.30% in LDS and 25.20% in SDS). AGR-Yr and CH-Yr data displayed a normal distribution, while in the CW and LSEN data followed a slightly bi-modal distribution (Figure 1). This was the most pronounced in CW data recorded in the LDS experiment. These data indicate the presence of sufficient variation for the four traits investigated, and show that the data is suitable to use in GWAS analysis.

Distribution of the phenotype of Canopy wilting (A), Leaf senescence (B), Drought index for the maximum absolute growth rate (C) and Drought index for the maximum canopy height (D). Different colors indicate the short duration stress experiment (SDS) and the long duration stress experiment (LDS)
Genome wide association analysis
The results of the GWAS analysis are shown in Additional file 1, and the Q-Q plots are presented in Additional file 2. GWAS identified 17 and 22 significant marker-trait associations (-logP ≥ 4) in the SDS and LDS experiments respectively (Figure 2). No single SNP-trait association was consistent over the two experiments, indicating that different genomic regions might be relevant for stress tolerance under short-duration and long-duration drought stress.

Manhattan plots of the GWAS analysis. The dashed horizontal line in blue indicates the threshold for -log10 P value ≥ 4. 2018 and 2019 in the plot’s titles represent ‘the short-duration stress experiment’ and ‘the long-duration stress experiment’ respectively
A total of 471 and 508 genes of interest were identified in the candidate regions delineated around the significant SNPs in the SDS and LDS experiments respectively (Additional file 3). A set of 12 and 16 of these genes (in the SDS and LDS experiment respectively) carried annotations for their functions of particular relevance for drought (Table 2). Furthermore, several previously reported QTLs targeting important traits, including some related to drought tolerance, co-located with the candidate regions (Table 2 and Additional file 4).
Canopy wilting (CW)
Ten SNPs displayed significant associations with CW in the SDS experiment (Table 2). These SNPs were distributed over seven chromosomes, and explained individually 2−8% of the phenotypic variation (R2). The candidate genes present in the candidate genomic regions surrounding the significantly associated SNPs are putatively involved in lateral root formation, growth, photosynthesis and cell-wall integrity in Arabidopsis plants (Table 2). For example, the candidate region defined by SNP AX-93918502 contains Glyma.03G261500, annotated as a P subunit of Photosystem-I protein encoding gene (PSI-P) and Glyma.03G258300, annotated as ‘Auxin Response Factor 18 (ARF18)’ which is known to regulate cell growth and seed weight in Arabidopsis. Glyma.03G261400, also present in the candidate region surrounding SNP AX-93918502, is annotated as ‘LEA hydroxyproline-rich family protein gene’, which plays a role in lateral root formation in Arabidopsis. Glyma.12G009100, in the candidate region defined by SNP AX-94092624 is an Arabidopsis homolog of ‘Reduced Lateral root Formation (RLF)’. The candidate region defined by SNP AX-93945641 contains Glyma.15G269200, an Arabidopsis homolog encoding a proline rich protein that has been shown to play a role in cell wall integrity and root elongation during drought stress in Arabidopsis. As shown in Table 2, SNP AX-93643614 collocates with an interesting QTL for water use efficiency (WUE).
For CW data recorded in the LDS experiment, five SNPs displayed significant associations. These SNPs were distributed over four chromosomes and explained individually 4-14% of the phenotypic variation (R2) for CW (Table 2). Among the genes identified in the candidate regions surrounding these SNPs, several are related to stress response, root formation, trans-membrane transport of water and solutes, stomatal movement, repair and maintenance of the photosynthetic apparatus and cellular protection through ROS scavenging. For example, the candidate region surrounding SNP AX-93976757 comprises Glyma.02G094700, annotated as ‘TIP1-3’ that putatively encodes a member of tonoplast intrinsic aquaporins (transmembrane channels for water and small uncharged solutes), Glyma.02G094900, annotated as ‘PsbP-like protein 1 (PLP1)’ which functions to repair damage in Photosystem II in Arabidopsis, and Glyma.02G097700, annotated as an ‘Aluminium activated malate transporter’, having a putative role in stomatal opening. The candidate region defined by SNP AX-93761082 contains Glyma.08G365700, an Arabidopsis orthologue of Abscisic acid-deficient 4 (ABA4) which is involved in stress-induced ABA accumulation. Interestingly, two QTLs for WUE have been also reported in this region (Table 2). Glyma.20G037100 (on Gm20) is an Arabidopsis orthologue of Lateral Root Primordium Protein-related (LRP1) and Glyma.20G197600 (on Gm20) is an Arabidopsis orthologue of an antioxidant protein gene ‘Glutathione S-transferase tau 4 (GSTU4)’.
Leaf senescence (LSEN)
Three SNPs locating on three different chromosomes displayed significant associations with LSEN in the SDS experiment, and explained up to 15% of the phenotypic variation. The candidate regions defined by these SNPs contain genes putatively involved in stress response, response to ABA and root cap development. The most strong effect was detected for a candidate region surrounding SNP AX-93917720, that contains Glyma.03G138000, which is annotated as a ‘Calcium-dependent Protein Kinase 19 (CPK8)’ known to be involved in ABA-mediated stomatal closure and ROS reduction in Arabidopsis plants. In the candidate region surrounding SNP AX-94142205, Glyma.15G266500 is annotated as ‘NAC (No Apical Meristem) domain transcriptional regulator’ (SMB), whose Arabidopsis orthologue controls root cap development. The region surrounding SNP AX-93886487 contains Glyma.19G027700, which is related to EID1-like 2, a member of F-box proteins and a positive regulator of ABA signaling to produce responses such as control of germination and root growth in Arabidopsis plants under drought stress conditions.
The 13 SNPs that displayed significant associations with LSEN in the LDS experiment are distributed over six chromosomes and explain 1-14% of the phenotypic variation. The candidate regions surrounding these SNPs contain genes involved in photosynthesis, ABA signaling, cellular repair and ROS scavenging. For example, the candidate region defined by SNP AX-93981961 contains Glyma.02G192700, annotated as ‘Calcium-dependent Protein Kinase 1 (CPK1)’, and Glyma.02G193000, related to ‘AtDjA3, a ‘DNAJ heat shock N-terminal domain-containing protein encoding gene’ in Arabidopsis. CPK1 functions in proline accumulation and ROS reduction, while AtDjA3 is known to maintain seed shape, seed size and seedling survival at high levels of ABA. The region between 25.5 Mb and 30 Mb on Gm16 contains four significant SNPs. A QTL (eChl_T) for total chlorophyll content has been reported for this region, and Glyma.16G145800, also present in this region is annotated as ‘LHCA1’ in Arabidopsis which encodes a photosystem I light harvesting complex chlorophyll a/b binding protein. The region between 48.1 Mb and 48.6 Mb on chromosome Gm18 contains seven significant SNPs, and contains two genes (Glyma.18G202900 and Glyma.18G203500) involved in salt and ABA responses and in ROS reduction. The candidate region defined by SNP AX-94195039 contains Glyma.19G191700 or ‘HSFA3’. In Arabidopsis, this gene encodes a heat shock protein that plays a role in thermotolerance and drought tolerance in combination with DREB2A.
Drought response for maximum absolute growth rate (AGRmax-Yr)
Only in the SDS experiment significant SNP-trait associations were found for AGRmax-Yr. These four SNPs were distributed over four chromosomes and explained up to 9% of the phenotypic variation. Genes putatively involved in stomatal movement, cell division, photosynthesis and response to ABA and stress response were contained in the delineated candidate regions. Glyma.05G128000 (orthologue of LHCB1.3 in Arabidopsis) putatively encodes a ‘light harvesting Photosystem II chlorophyll a/b binding protein’ that regulates stomatal movement and plant responses to drought, while Glyma.18G259700 is an ortholgoue of FtsH protease that functions in thylakoid membrane biogenesis and Photosystem II repair in Arabidopsis. On Gm06, Glyma.06G204100 is annotated as ‘ABI5 binding protein 3 (AFP3)’ which negatively regulates ABA response in Arabidopsis. Two previously reported QTLs for WUE are also present in the same region on Gm06. In addition, the same region is located in the neighborhood (at 4.1 Mb) of one candidate region for CW (CW_SDS_5) (Table 2).
Drought response for maximum canopy height (CH-Yr)
For CH-Yr, significant SNP-trait associations were only detected in the LDS experiment. In this case four SNPs distributed over four chromosomes and explaining 6-16% of the variation displayed significant associations. The candidate genes identified in the neighborhood of these significant SNPs are putatively involved in stress response, stomatal movement and photosynthesis. Glyma.06G202200 is orthologue to ‘HSP101’ which encodes a heat shock protein in Arabidopsis. On Gm08, Glyma.08G191300 is annotated as ‘calmodulin-binding family protein gene’ which is known to be involved in stomatal opening in Arabidopsis. On Gm09, Glyma.09G071400 (or ‘LHCA6’ in Arabidopsis) encodes a ‘light harvesting Photosystem I chlorophyll a/b binding protein 6’. A previously reported QTL for WUE is also present in the same region on Gm09.
Some candidate regions for CH-Yr are located close to candidate regions for CW and LSEN. i.e. on Gm16 CH-Yr_LDS_4 is located next to LSEN_LDS_3, LSEN_LDS_4a and LSEN_LDS_ 4b with a maximum distance of 2.6 Mbp between them, and on Gm16, CH-Yr_LDS_1 is located next to CW_SDS_5 with 3.6 Mbp between them (Table 2).
Discussion
GWAS revealed novel loci associated with tolerance to a short duration and a long duration drought stress
The GWAS analysis identified a total of 17 and 22 significant marker-trait associations for four traits (CW, LSEN, AGRmax-Yr, CH-Yr) in the short-duration and long-duration drought experiments respectively. None of these SNPs was common to the two experiments. This was not completely unexpected because the overall phenotypic response of the EUCLEG soybean collection in two experiments was different [23]. A total of 12 and 16 of the genes that are in LD with these SNPs (for SDS and LDS experiments respectively) are of particular relevance for drought stress responses including stomatal movement, root formation, photosynthesis, ABA signaling, cellular protection and cellular repair mechanisms. Furthermore multiple previously reported QTLs related to drought tolerance traits such as WUE, chlorophyll content and photosynthesis, and QTLs for traits including plant height and other yield-related traits, disease tolerance, nutrient content, seed composition also co-localize with the significantly associated SNPs.
Short duration and long duration drought treatment induce differential mechanisms of canopy wilting in soybean
Slow or delayed wilting is known to be a complex trait, that can be the result of several underlying mechanisms in soybean: (i) good water resource exploration by a large root system [101]; (ii) lower stomatal conductance, reduced transpiration rate and high water use efficiency [102]; (iii) maintenance of constant transpiration rate under vapor pressure deficit conditions above 2.0 kPa [103]; and (iv) lower radiation use efficiency [104]. Several of the candidate genes identified in the SDS experiment are putatively involved in lateral root development, and some are related to photosynthesis. Root system modification is an important response that shows a large phenotypic plasticity in soybean under drought stress [15]. This response is a drought avoidance mechanism enabling plants to sustain high plant water status or cellular hydration under drought [105]. Drought stress inhibits root growth in general but susceptible genotypes show more prominent effects [106, 107] due to an overall decrease in newly synthesized cell wall polysaccharides such as pectin, hemicellulose, and cellulose [108]. [107] discuss how the taproot length and tertiary root length influence the root surface area, what in turn influences the plant nutrient and water absorption capacity. Similarly, soybean genotypes with high root length, surface area, diameter and volume achieved high net photosynthesis, attained higher plant height and biomass, and tended to perform better under water deficit conditions [106]. The well-described slow wilting soybean accession PI 796397 has a dense root system with a high number of root tips [109]. We have shown a large level of variation for drought index for canopy height (CH-Yr) and number of pods per main stem (PPS-Yr) in the EUCLEG soybean collection [23]. Some accessions were able to maintain high CH and displayed no reduction in the number of pods per plant (NPP), but the relationship between these traits and CW was not clear. This might be due to compensation during the period of stress recovery as discussed in [23]. Anyhow, the results obtained in this work regarding marker-trait associations support the hypothesis that the mechanism of slow wilting in the SDS experiment might be associated with the characteristics of the root system.
In the LDS experiment, the associated SNPs were in LD with genes related to stress-induced abscisic acid (ABA) accumulation, stability of the photosynthetic apparatus, oxidative stress response and ROS scavenging, and trans-membrane water and solute transport, along with one gene related to root development. Some candidate regions in LDS also overlapped with previously reported QTLs for WUE [110]. In [23] we have reported a drastic reduction of the growth rate, plant height and yield in the LDS treatment. These plant responses match with the dehydration tolerance mechanism that is activated under extreme drought stress conditions [111]. Dehydration tolerant plants tend to maintain metabolic activities at low tissue water potential through osmotic adjustment, antioxidant activities, and altered growth regulators [112]. Soybean plants subjected to drought for 20 days at V5 stage in [113] showed a higher accumulation of ABA, lower stomatal conductance and decreased rate of net photosynthesis, but resistant genotypes performed relatively better than the susceptible ones. In [114], drought stress disturbed the balance between ROS and antioxidant enzymes, and a severe stress led to overproduction of ROS causing cellular damage, low stomatal conductance and a decrease in photosynthesis parameters in soybean. Drought tolerant soybean genotypes in [22] developed an anti-oxidative defense mechanism for ROS scavenging by increasing the activity of antioxidant enzymes including the peroxidase superoxide dismutase, ascorbate peroxidase, glutathione reductase and catalase, which helped them to maintain a high photosynthetic efficiency and a high RuBisCo activity under heavy stress conditions. Our results suggest that under severe stress, canopy wilting response in soybean is regulated by a different mechanism than under short duration stress. Based on the marker-traits associations identified in the LDS experiment, slow wilting genotypes can achieve a high WUE and thus a high level of tolerance to long-term drought stress through a reduction of stomatal conductance and transpiration rates.
Drought-induced leaf senescence is related to ABA and ROS responses in soybean
GWAS identified 3 and 13 significant SNPs for LSEN in the SDS and LDS experiments respectively. Premature leaf senesce is thought to be an indicator of stress vulnerability in plants [115]. [116] described a negative correlation between premature leaf senescence and plant survival in a perennial temperate grass. According to the mechanism of premature leaf senescence, accumulation of ABA under stress conditions promotes overproduction of ROS which may react with proteins, lipids and deoxyribonucleic acid, leading to oxidative damage and premature leaf senescence [84, 85]. Several transcription factors that play a role in the control of age-induced leaf senescence, play also a role in plant stress tolerance, and chloroplast breakdown is a common feature in both cases [115]. Here we also identified multiple candidate genes for LSEN. Three candidate genes known to be involved in ABA-mediated stomatal closure, root cap development and ROS scavenging were identified in the SDS experiment. In the LDS experiment, six candidate genes had stress related functions such as ABA-mediated seedling survival, maintenance of seed shape and seed size at high ABA levels, drought and salt tolerance through proline accumulation and ROS scavenging, and stability of Photosystem I. In the work of [117] a higher WUE and an increased tolerance obtained through the activation of a cell wall invertase in tomato was associated with a low stomatal conductance, delayed senescence, increased source activity and a better control of ROS production under drought stress. In [118] minimizing the stress-mediated senesce by the over expression of a senescence associated gene enabled the plants to attain higher shoot and root biomass and to recover better after a period of drought stress. Our results also reveal that drought-induced premature senescence in soybean can be avoided by targeting the genes involved in ROS scavenging and stomatal conductance.
Furthermore, our results reveal that two candidate regions (LSEN_LDS_3 and LSEN_LDS_4a on Gm16) can be of particular significance as they co-localize with QTLs for net photosynthesis rate (qP16; [63]) and for total chlorophyll content (eChl-T; [62]). Moreover, the candidate gene Glyma.16G145800, having a putative role in chlorophyll synthesis [87], is also present in the candidate region LSEN_LDS_4a. It has been discussed that the drought-induced accelerated senescence affects source-sink relationship in wheat [119] and causes a significant yield reduction in soybean [120]. The candidate region LSEN_LDS_6 on Gm19 contains a gene encoding a heat shock protein which confers stress tolerance [91, 92]. The same region also contains many previously reported QTLs for important agronomic traits including leaf growth, plan height, internode length, number of pods and seed amino acid content, indicating the relevance of this candidate region to improve drought tolerance and agronomic performance in soybean.
QTLs related to WUE play a role in the maintenance of growth rate and canopy height under drought stress
Our GWAS analysis identified also candidate genes for drought response of maximum absolute growth rate (AGRmax-Yr) and drought response of maximum canopy height (CH-Yr). These traits were determined using UAV-based methods [23], and quantify the growth of the soybean accessions in the drought treatment relative to the control treatment. AGRmax-Yr and CH-Yr are therefore comprehensive indicators of drought tolerance, and are derived by the actions of multiple physiological processes, including the rate of carbon assimilation, photosynthesis, respiration, source and sink relations, nutrient balances, cell differentiation and elongation, as well as belowground processes [43, 121, 122]. A total of four significant associations were identified by GWAS for AGRmax-Yr in the SDS experiment, and another four significant associations for CH-Yr in the LDS experiment. Allelic differences of the significant SNPs explained a low to high drought-induced reduction for maximum absolute growth rate and for maximum canopy height in the EUCLEG accessions. The comprehensive nature of these growth related traits was reflected in the fact that some of the candidate regions for AGRmax-Yr and CH-Yr are located closely to the other candidate regions for CW and LSEN (on Gm05, Gm06 and Gm16; Table 2).
Furthermore, two previously reported QTLs for WUE [110] coincided with the candidate region AGRmax-Yr_SDS_2, on Gm06. This region contains Glyma.06G204400, whose Arabidopsis orthologue TRM4 or LONGIFOLIA is considered essential for cellulose deposition [95], whereas cellulose is essential for stem growth [96]. The same region contains Glyma.06G204100, annotated as Arabidopsis AFP3, which is a negative regulator of ABA response as it causes proteomic degradation of ABI5 [94], a core element in ABA response [123]. Furthermore, another candidate region CH-Yr_LDS_1, on Gm06 contains Glyma.06G202200, annotated as Heat-shock Protein 101 in Arabidopsis (HSP101). Constitutive expression of HSP101 provided heat tolerance and a high survival rate without any detrimental effect on growth in Arabidopsis plants [98]. These two regions on Gm06 can be of particular interest for further research and to modulate drought tolerance in soybean.
SNPs in candidate regions CH-Yr-LDS_2, _3 and _4 explained a relatively large proportion of the phenotypic variation for CH-Yr (R2 = 0.16; Table 2). The candidate gene Glyma.08G191300 (Arabidopsis EDA39) on Gm08 belongs to the calmodulin-binding family protein. EDA39, in coordination with AtWRKY21, promotes stomatal opening by down-regulating the ABA response [124]. Another interesting candidate gene is Glyma.09G071400 (Arabidopsis LHCA6), present on Gm09. LHCA6 was shown to play a role in the normal functioning of the chloroplast [125], and its reduced expression caused a lower photosynthesis in Arabidopsis plants [100].
Considering the high level of stress imposed in the LDS treatment, and the results presented above for canopy wilting in the LDS experiment, it is obvious that within the EUCLEG collection some accessions have mechanisms to minimize transpiration through stomatal regulation, ensuring survival under long-term stress treatment. Therefore, our results suggest that a high water use efficiency could be an effective means to improve drought tolerance of this European soybean collection, in particular for environments in which the chance of long periods of drought is high. Further, our results indicate that for an effective drought tolerance under long-term drought stress, modulating the genes related to photosynthesis could be promising in soybean.
Conclusion
The present study applied a genome-wide association study (GWAS) analysis and investigated the genetic control of drought tolerance in the EUCLEG soybean collection relevant for breeding in Europe. The GWAS analysis identified a total of 17 and 22 significant marker-trait associations for four traits, in the short duration drought (SDS) and long duration drought (LDS) experiments, respectively. Based on these marker-trait associations a total of 12 and 16 genes of particular relevance for drought stress responses including the stomatal movement, root formation, photosynthesis, ABA signaling, cellular protection and cellular repair mechanisms, were identified. Several previously known QTLs for drought tolerance traits such as WUE, chlorophyll content and photosynthesis, co-localized with the significantly associated SNPs, suggesting their role in drought tolerance mechanisms in soybean. Our results revealed a differential mechanism of canopy wilting in the EUCLEG collection under SDS and LDS. The marker-trait associations in the SDS targeted multiple genes involved in root elongation, root cap development, lateral root formation etc., suggesting that slow canopy wilting in the SDS might be associated with the characteristics of root system. In the LDS, several of identified genes and previously known QTLs carried annotations related to water transport, maintenance of photosynthetic apparatus and cellular protection and WUE, suggesting that slow wilting response under the LDS might be associated with a high WUE through a reduction of stomatal conductance and transpiration rates. Our results also showed that the drought-induced leaf senescence could be related to ABA and ROS responses in soybean, and QTLs related to WUE might have played a role in the determination of growth rate and canopy height under drought stress. Many previously reported QTLs for multiple agronomic traits also co-localized with the significantly associated SNPs, suggesting that the identified genomic regions might have the potential to improve the agronomic performance together with the drought tolerance in the EUCLEG collection. These results provide an important basis to improve drought tolerance in soybean in Europe. The genomic regions identified in this study can be further explored to ensure their added value in the genomic assisted breeding i.e. genomic selection.
Materials and methods
Field trials and phenotypic evaluation
The set of 359 soybean accessions originating from 25 countries in Europe, China and the USA of [23] was used. This set is a part of the EUCLEG collection assembled in context of the EUCLEG project (www.eucleg.eu). It represents widely the germplasm available for breeding of soybean in Europe. They are the gene bank accessions from the ARS-GRIN database and the IPK database, and the accessions from the collaborators of the EUCLEG project. This set has been described in [23] and [64]. The accessions were divided into four groups (GP), based on maturity information [23]. Accessions in GP1 (n=90), GP2 (n=91), GP3 (n=88), and GP4 (n=90) were expected to belong to maturity groups MGI/II, MG0, MG00, and MG000, respectively [23]. This wide representation of genetic diversity of the EUCLEG collection makes it an ideal collection for evaluating its response to drought stress and subsequently for the GWAS analysis to identify the genomic loci affecting the drought tolerance of soybean.
These accessions were sown in 2018 and 2019 in Melle, Belgium (51.00° N, 3.80° E) on sandy loam soil. The experimental set-up has already been described in [23]. In short, each year (2018 and 2019) two adjacent fields sown. One of these fields was used as ‘control’, while the other was subjected to a ‘drought’ treatment. The four GPs were sown on four different dates (between 20 April and 11 May in 2018 and between 19 April and 15 May in 2019) to synchronize the developmental stage at which drought was imposed (i.e. when 50% of the plots had initiated flowering). The plot dimensions were slightly adjusted for four different GPs in order to implement their recommended row-to-row spacing and sowing densities (45, 55, 65, and 75 seeds m-2 for GP1, GP2, GP3, and GP4, respectively). Each plot contains three rows. For GP1 and GP2 with a row-to-row spacing of 0.40 m, plot dimensions were 1.20 x 0.75 m with (area 0.90 m2). For GP3 and GP4 with a row-to-row spacing of 0.25 m, plot dimensions were 0.75 x 1.20 m. Irrigation was applied to maintain sufficient soil moisture in the drought and control fields until flowering had started on 50% of the plots. After that, a period of drought was applied for 3-4 weeks in 2018, and for 6-7 weeks in 2019 to the plots of the drought field. This was achieved by placing mobile rain-out shelters. The irrigation was continued in the control field. After the drought treatment, irrigation was resumed in the drought field and was continued until the late development in control and drought fields.
We evaluated canopy wilting (CW) and leaf senescence (LSEN) symptoms during the period of drought treatment in the drought fields, as described in [23]. CW was scored (score 1-9) three times in 2018 and four times in 2019. LSEN was scored (score 1-9) during and at the end of the drought period. UAV based methodology [126] was used to determine canopy height in each plot using RGB cameras mounted on drones. A growth curve was fitted to the data, and the values of maximum absolute growth rate (AGRmax) and maximum canopy height (CH) were derived for each accession in control and drought fields.
Phenotypic data analysis
To proceed with the data analysis, filtering was first applied. Plots with less than 30% seedling emergence and outliers identified using Tukey’s rule [127] were removed from the datasets to avoid any bias in analysis. This filtering criteria for seedling emergence was considered sufficient, knowing that on average the emergence was 54-68% in control-drought treatments considering both years 2018 and 2019 (Additional file 1). Data of 2018 and 2019 were considered separately (see [23] for more details). The filtered and cleaned data were analyzed using mixed linear models with the lmer() function integrated in the lme4 package in R [128] using the following base model:
Where, ‘Y’ is response variable, ‘Intercept’ represents the overall mean value of the response variable. ‘Genotype’ is a random effect representing the accession, and ‘Block’, ‘Column’ and ‘Row’ are random effects representing spatial components in the experimental design. ‘Residual’ represents the noise term. The random effects for Block, Column and Row were assumed to be independent, and to originate from an identical, normal distribution. The residuals were also assumed to be independent and identically distributed. lmer() uses restricted maximum likelihood (REML) to estimate the variance parameters of the random components (i.e. Genotype, Block, Column and Row).
For each response variable six versions of the base model were tested (considering different combinations of effects) and the output was evaluated using the Akaike Information Criterion (AIC) [129]. The best model was then chosen based on the lowest AIC value. The following choices were made, as reported in [23]:
CW 2018, CW 2019, LSEN 2019, AGRmax 2018, AGRmax 2019, CH 2018, CH 2019:
Y = Intercept + Genotype + Column + Row + Residual
LSEN 2018:
Y= Intercept + Genotype + Row + Residual
The best linear unbiased predictor value (BLUP) was then calculated for each accession as the sum of the ‘Intercept’ value and the value of the random effect of ‘Genotype’.
A drought index (Yr) was calculated for the CH and AGRmax data according to [130]:
Where, \({\widehat{\mu }}_{c}\) and \({\widehat{\mu }}_{D}\) are the estimated BLUPs values of the traits from control and drought treatments respectively. Yr quantifies the strength of the response of each accession to drought stress.
SNP data
The SNP data included in the present study has been described in [64], where a full description of the methods for genotyping and SNP calling is provided. In short, a total of 477 EUCLEG soybean accessions (including the 359 accessions considered in the present study) were genotyped using the NJAU 350KSoySNP microarray [131]. The same microarray was also used to genotype 394 accessions from the NJAU collection from China. To minimize the chance of misclassification of SNPs, which reduces the false positives in the subsequent analysis, SNP calling was performed on the genotyping data of the EUCLEG and NJAU collections together. The joint analysis provided a total 224,993 SNP markers of high quality spread over the 20 soybean chromosomes with an average distance of 2.6 Kbp between two adjacent SNPs. For the present study we extracted the SNP loci that were polymorphic in the set of 359 EUCLEG accessions. This rendered a set of 139,427 high quality SNPs which are polymorphic at MAF ≥ 0.05.
Association mapping
The GWAS analysis was performed separately for the SDS and LDS experiments with a Bayesian-information and Linkage-disequilibrium Iteratively Nested Keyway model (BLINK) [132] implemented in the GAPIT R package [133]. BLINK was used with the default settings and LD = 0.2. A threshold value of -Log10 P-value ≥ 4 was set to declare a marker-trait association as significant. The variance explained by each SNP (R2) was calculated by applying a simple linear regression between SNP genotype and the phenotype data using lm() function in R 3.6.3 [134]. The allele substitution effect of each SNP was calculated by taking the difference in the median value between genotypes carrying the major allele and the minor allele.
Identification of candidate genes
Candidate regions were delineated around the significantly associated SNPs based on the Linkage disequilibrium (LD) decay distance per chromosome as reported in [64]. LD decay distance per chromosome was calculated in the full EUCLEG collection (477 accessions including the 359 accessions of the present study) and using 224,993 SNPs (including the subset of 139,427 SNPs used in the present study). Candidate regions refer then to the genomic regions surrounding the significantly associated SNPs and spanning up to the LD decay distance calculated for the chromosome where the corresponding SNP is located. A list of genes (genes of interest) located in these candidate regions was retrieved from the soybean reference genome Wm82.a2.v1 using [135]. The corresponding orthologous Arabidopsis thaliana genes and their functional annotation were obtained from [65]. [63, 66] and [62] were used to retrieve previously reported QTLs in the candidate regions.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Abbreviations
- ABA:
-
Abscisic acid
- AGRmax-Yr:
-
Drought index for maximum absolute growth rate
- BLINK:
-
Bayesian-information and Linkage-disequilibrium Iteratively Nested Keyway model
- BLUPs:
-
Best Linear Unbiased Predictors
- CH-Yr:
-
Drought index for maximum canopy height
- CW:
-
Canopy wilting
- GWAS:
-
Genome-wide association study
- GST:
-
Glutathione-S-Transferase
- LDS:
-
Long duration drought stress experiment
- LEA:
-
Late Embryogenesis Abundant
- LSEN:
-
Leaf senescence
- NDVI:
-
Normalized Difference Vegetation Index
- SDS:
-
Short duration drought stress experiment
- SNPs:
-
Single Nucleotide Polymorphisms
- QTLs:
-
Quantitative Trait Loci
- Rab:
-
Response to abscisic acid
- ROS:
-
Reactive Oxygen Species
- WSC:
-
Water-soluble carbohydrates
- WUE:
-
Water use efficiency
References
Mbow C, Rosenzweig C, Barioni LG, Benton TG, Herrero M, Krishnapillai M, et al. Food security. In: Shukla PR, Skea J, Buendia E. C, Masson-Delmotte V, Pörtner HO, Roberts DC, et al., editors. Climate Change and Land: an IPCC special report on climate change, desertification, land degradation, sustainable land management, food security, and greenhouse gas fluxes in terrestrial ecosystems. In press; 2019 [accessed 2022 Feb 23]. https://www.ipcc.ch/site/assets/uploads/sites/4/2021/02/08_Chapter-5_3.pdf.
FAO. The future of food and agriculture Trends and Challenges. Rome; 2017 [accessed 2021 Feb 8]. http://www.fao.org/3/i6583e/i6583e.pdf.
Lesk C, Rowhani P, Ramankutty N. Influence of extreme weather disasters on global crop production. Nat. 2016;529(7584):84–7. https://www.nature.com/articles/nature16467.
Luo L, Xia H, Lu BR. Editorial: Crop breeding for drought resistance. Front Plant Sci. 2019;10:314.
Mundhe S, Patil R, Oak M, Jaybhay S. Accelerating Soybean Improvement Through Genomics-Assisted Breeding. In: Accelerated Plant Breeding, Volume 4 Oil Crops. Springer Nature, Switzerland AG.; 2022. p. 41–62. https://doi.org/10.1007/978-3-030-81107-5.
Sgroi LC, Lovino MA, Berbery EH, Müller GV. Characteristics of droughts in Argentina’s core crop region. Hydrol Earth Syst Sci. 2021;25(5):2475–90. https://doi.org/10.5194/HESS-25-2475-2021.
Wang C, Linderholm HW, Song Y, Wang F, Liu Y, Tian J, et al. Impacts of Drought on Maize and Soybean Production in Northeast China During the Past Five Decades. Int J Environ Res Public Health. 2020;17(7):2459. https://doi.org/10.3390/IJERPH17072459.
FAS USDA. World Agricultural Production. 2022 [accessed 2022 Feb 24]. https://apps.fas.usda.gov/psdonline/circulars/production.pdf.
Engels C, Rodrigues FA, Ferreira A de O, Inagaki TM, Nepomuceno AL. Drought effects on soybean cultivation - A review. Annu Res Rev Biol. 2017;16(1).
Arya H, Singh MB, Bhalla PL. Towards Developing Drought-smart Soybeans. Front Plant Sci. 2021;12:2123. https://doi.org/10.3389/FPLS.2021.750664.
Wang Z, Li G, Sun H, Ma L, Guo Y, Zhao Z, et al. Effects of drought stress on photosynthesis and photosynthetic electron transport chain in young apple tree leaves. Biol Open. 2018;7(11). https://pubmed.ncbi.nlm.nih.gov/30127094/.
Ahmad P, Jamsheed S. Hameed A, Rasool S, Sharma I, Azooz MM, et al. Drought Stress Induced Oxidative Damage and Antioxidants in Plants. In P. Ahmad (Ed.). Oxidative Damage to Plants 2014. (pp.345–367). Elsevier Inc. https://doi.org/10.1016/B978-0-12-799963-0.00011-3.
Kapoor D, Bhardwaj S, Landi M, Sharma A, Ramakrishnan M, Sharma A. The Impact of Drought in Plant Metabolism: How to Exploit Tolerance Mechanisms to Increase Crop Production. Appl Sci 2020;10(16):5692. https://www.mdpi.com/2076-3417/10/16/5692/htm.
Jorge JG, Villalobos-López MA, Chavarría-Alvarado KL, Ríos-Meléndez S, López-Meyer M, Arroyo-Becerra A. Genome-wide transcriptional changes triggered by water deficit on a drought-tolerant common bean cultivar. BMC Plant Biol. 2020;20(1):1–20. https://bmcplantbiol.biomedcentral.com/articles/https://doi.org/10.1186/s12870-020-02664-1.
Ye H, Roorkiwal M, Valliyodan B, Zhou L, Chen P, Varshney RK, et al. Genetic diversity of root system architecture in response to drought stress in grain legumes. J Exp Bot. 2018;69(13):3267–77. https://academic.oup.com/jxb/article/69/13/3267/4924018.
Tardieu F, Simonneau T, Muller B. The Physiological Basis of Drought Tolerance in Crop Plants: A Scenario-Dependent Probabilistic Approach. Annu Rev Plant Biol. 2018;69:733–59. https://pubmed.ncbi.nlm.nih.gov/29553801/.
Keneni G, Bekele E, Imtiaz M, Dagne K, Alemaw G. Challenges Associated with Crop Breeding for Adaptation to Drought-Prone Environments. Ethiop J Agric Sci. 2017;27(1):1–24. https://www.ajol.info/index.php/ejas/article/view/150327.
Gupta PK, Balyan HS, Gahlaut V. QTL Analysis for Drought Tolerance in Wheat: Present Status and Future Possibilities. Agron. 2017;7(1):5. https://www.mdpi.com/2073-4395/7/1/5/htm.
Gupta A, Rico-Medina A, Caño-Delgado AI. The physiology of plant responses to drought. Science. 2020;368(6488):266–9. https://doi.org/10.1126/science.aaz7614.
Zhao T, Aleem M, Sharmin RA. Adaptation to Water Stress in Soybean: Morphology to Genetics. In: Andjelkovic V, editor. Plant, Abiotic Stress and Responses to Climate Change. IntechOpen 2017. p 186. https://www.intechopen.com/chapters/58038.
Dong S, Jiang Y, Dong Y, Wang L, Wang W, Ma Z, et al. A study on soybean responses to drought stress and rehydration. Saudi J Biol Sci. 2019;26(8):2006–17.
Iqbal N, Hussain S, Raza MA, Yang CQ, Safdar ME, Brestic M, et al. Drought Tolerance of Soybean (Glycine max L. Merr.) by Improved Photosynthetic Characteristics and an Efficient Antioxidant Enzyme Activities Under a Split-Root System. Front Physiol. 2019;10:786. https://doi.org/10.3389/FPHYS.2019.00786.
Saleem A, Aper J, Muylle H, Borra-Serrano I, Quataert P, Lootens P, et al. Response of a Diverse European Soybean Collection to “Short Duration” and “Long Duration” Drought Stress. Front Plant Sci. 2022;13:818766. https://doi.org/10.3389/fpls.2022.818766.
Wei Y, Jin J, Jiang S, Ning S, Liu L. Quantitative Response of Soybean Development and Yield to Drought Stress during Different Growth Stages in the Huaibei Plain, China. Agron. 2018;8(7):97. https://www.mdpi.com/2073-4395/8/7/97/htm.
Wu X, Sun T, Xu W, Sun Y, Wang B, Wang Y, et al. Unraveling the Genetic Architecture of Two Complex, Stomata-Related Drought-Responsive Traits by High-Throughput Physiological Phenotyping and GWAS in Cowpea (Vigna. Unguiculata L. Walp). Front Genet. 2021;12:2158.
Teran JCBM y, Konzen ER, Palkovic A, Tsai SM, Rao IM, Beebe S, et al. Effect of drought stress on the genetic architecture of photosynthate allocation and remobilization in pods of common bean (Phaseolus vulgaris L.), a key species for food security. BMC Plant Biol. 2019;19(1):1–15. https://doi.org/10.1186/s12870-019-1774-2.
Muchero W, Roberts PA, Diop NN, Drabo I, Cisse N, Close TJ, et al. Genetic architecture of delayed senescence, biomass, and grain yield under drought stress in cowpea. PLoS One. 2013;8(7). https://pubmed.ncbi.nlm.nih.gov/23936140/.
Nezhadahmadi A, Prodhan ZH, Faruq G. Drought Tolerance in Wheat. Rajakaruna N, Boyd RS, editors. Sci World J. 2013. https://doi.org/10.1155/2013/610721.
Jain N, Singh GP, Singh PK, Ramya P, Krishna H, Ramya KT, et al. Molecular approaches for wheat improvement under drought and heat stress. Indian J Genet Plant Breed. 2014;74(4):578–83. https://doi.org/10.5958/0975-6906.2014.00893.1.
Liu S, Qin F. Genetic dissection of maize drought tolerance for trait improvement. Mol Breed. 2021;41(2):1–13. https://doi.org/10.1007/s11032-020-01194-w.
Ribaut JM, Ragot M. Marker-assisted selection to improve drought adaptation in maize: the backcross approach, perspectives, limitations, and alternatives. J Exp Bot. 58(2):351–60. https://pubmed.ncbi.nlm.nih.gov/17158111/.
Rasheed A, Hassan MU, Aamer M, Batool M, Fang S, Wu Z, et al. A critical review on the improvement of drought stress tolerance in rice (Oryza sativa L.). Not Bot Horti Agrobot Cluj-Napoca. 2020;48(4):1756–88. https://www.notulaebotanicae.ro/index.php/nbha/article/view/12128.
Swamy BPM, Ahmed HU, Henry A, Mauleon R, Dixit S, Vikram P, et al. Genetic, physiological, and gene expression analyses reveal that multiple QTL enhance yield of rice mega-variety IR64 under drought. PLoS One. 2013;8(5). https://pubmed.ncbi.nlm.nih.gov/23667521/.
Ramayya PJ, Vinukonda VP, Singh UM, Alam S, Venkateshwarlu C, Vipparla AK, et al. Marker-assisted forward and backcross breeding for improvement of elite Indian rice variety Naveen for multiple biotic and abiotic stress tolerance. PLoS One. 2021;16(9):e0256721. https://doi.org/10.1371/journal.pone.0256721.
Charlson D V., Bhatnagar S, King CA, Ray JD, Sneller CH, Carter TE, et al. Polygenic inheritance of canopy wilting in soybean [Glycine max (L.) Merr.]. Theor Appl Genet. 2009;119(4):587–94. https://pubmed.ncbi.nlm.nih.gov/19471903/.
Abdel-Haleem H, Carter TE, Purcell LC, King CA, Ries LL, Chen P, et al. Mapping of quantitative trait loci for canopy-wilting trait in soybean (Glycine max L. Merr). Theor Appl Genet. 2012;125(5):837–46. https://pubmed.ncbi.nlm.nih.gov/22566068/.
Hwang S, King CA, Chen P, Ray JD, Cregan PB, Carter TE, et al. Meta-analysis to refine map position and reduce confidence intervals for delayed-canopy-wilting QTLs in soybean. Mol Breed. 2016;36(7):1–14. https://doi.org/10.1007/s11032-016-0516-5.
Ye H, Song L, Schapaugh WT, Ali ML, Sinclair TR, Riar MK, et al. The importance of slow canopy wilting in drought tolerance in soybean. J Exp Bot. 2020;71(2):642–52. https://academic.oup.com/jxb/article/71/2/642/5450730.
Kaler AS, Ray JD, Schapaugh WT, King CA, Purcell LC. Genome-wide association mapping of canopy wilting in diverse soybean genotypes. Theor Appl Genet. 2017;130(10):2203–17. https://doi.org/10.1007/s00122-017-2951-z.
Chamarthi SK, Kaler AS, Abdel-Haleem H, Fritschi FB, Gillman JD, Ray JD, et al. Identification and Confirmation of Loci Associated With Canopy Wilting in Soybean Using Genome-Wide Association Mapping. Front Plant Sci. 2021;12:698116. https://doi.org/10.3389/FPLS.2021.698116.
Bazzer SK, Purcell LC. Identification of quantitative trait loci associated with canopy temperature in soybean. Sci. Reports 2020;10(1):1–11. https://www.nature.com/articles/s41598-020-74614-8.
Kaler AS, Ray JD, Schapaugh WT, Asebedo AR, King CA, Gbur EE, et al. Association mapping identifies loci for canopy temperature under drought in diverse soybean genotypes. Euphytica. 2018;214(8):1–18. https://doi.org/10.1007/s10681-018-2215-2.
Wang W, Zhou B, He J, Zhao J, Liu C, Chen X, et al. Comprehensive Identification of Drought Tolerance QTL-Allele and Candidate Gene Systems in Chinese Cultivated Soybean Population. Int J Mol Sci. 2020;21(14):4830. https://www.mdpi.com/1422-0067/21/14/4830/htm.
Dhungana SK, Park JH, Oh JH, Kang BK, Seo JH, Sung JS, et al. Quantitative Trait Locus Mapping for Drought Tolerance in Soybean Recombinant Inbred Line Population. Plants 2021;10(9):1816. https://www.mdpi.com/2223-7747/10/9/1816/htm.
Hu Z, Zhang D, Zhang G, Kan G, Hong D, Yu D. Association mapping of yield-related traits and SSR markers in wild soybean (Glycine soja Sieb. and Zucc.). Breed Sci. 2014;63(5):441. https://doi.org/10.1270/JSBBS.63.441.
Hao D, Cheng H, Yin Z, Cui S, Zhang D, Wang H, et al. Identification of single nucleotide polymorphisms and haplotypes associated with yield and yield components in soybean (Glycine max) landraces across multiple environments. Theor Appl Genet. 2012;124(3):447–58. https://pubmed.ncbi.nlm.nih.gov/21997761/.
Contreras-Soto RI, Mora F, Oliveira MAR de, Higashi W, Scapim CA, Schuster I. A Genome-Wide Association Study for Agronomic Traits in Soybean Using SNP Markers and SNP-Based Haplotype Analysis. PLoS One. 2017;12(2):e0171105. https://doi.org/10.1371/journal.pone.0171105.
Kurasch AK, Hahn V, Leiser WL, Vollmann J, Schori A, Bétrix CA, et al. Identification of mega-environments in Europe and effect of allelic variation at maturity E loci on adaptation of European soybean. Plant Cell Environ. 2017;40(5):765–78. https://pubmed.ncbi.nlm.nih.gov/28042879/.
European Commission. REPORT FROM THE COMMISSION TO THE COUNCIL AND THE EUROPEAN PARLIAMENT on the development of plant proteins in the European Union. 2018 [accessed 2021 Jan 27]. https://ec.europa.eu/commission/presscorner/detail/en/IP_18_6495.
FAOSTAT. Food and Agriculture Organization of the United Nations (FAO). FAOSTAT Database. 2021 [accessed 2021 Jan 11]. http://faostat.fao.org/site/291/default.aspx.
IDH, IUCN NL. EUROPEAN SOY MONITOR. 2019 [accessed 2019 Nov 23]. https://www.idhsustainabletrade.com/uploaded/2019/04/European-Soy-Monitor.pdf.
Boulch G, Elmerich C, Djemel A, Lange B. Evaluation of soybean (Glycine max L.) adaptation to northern European regions under different agro-climatic scenarios. in silico Plants. 2021;3(1):1–13. https://academic.oup.com/insilicoplants/article/3/1/diab008/6128836.
Fu Z, Ciais P, Bastos A, Stoy PC, Yang H, Green JK, et al. Sensitivity of gross primary productivity to climatic drivers during the summer drought of 2018 in Europe. Philos Trans R Soc B. 2020;375(1810). https://doi.org/10.1098/rstb.2019.0747.
Bastos A, Fu Z, Ciais P, Friedlingstein P, Sitch S, Pongratz J, et al. Impacts of extreme summers on European ecosystems: a comparative analysis of 2003, 2010 and 2018. Philos Trans R Soc B. 2020;375(1810):20190507. https://doi.org/10.1098/rstb.2019.0507.
Peters W, Bastos A, Ciais P, Vermeulen A. A historical, geographical and ecological perspective on the 2018 European summer drought. Philos Trans R Soc B. 2020;375(1810). https://doi.org/10.1098/rstb.2019.0505.
UNDRR. United Nations Office for Disaster Risk Reduction. GAR Special Report on Drought 2021. Geneva; 2021 [accessed 2021 Sep 8]. https://www.undrr.org/publication/gar-special-report-drought-2021.
Kranz WL, Specht JE. Irrigating soybean. NebGuide G1367 2012. http://ianrpubs.unl.edu/epublic/live/g1367/build/g1367.pdf.
Yan C, Song S, Wang W, Wang C, Li H, Wang F, et al. Screening diverse soybean genotypes for drought tolerance by membership function value based on multiple traits and drought-tolerant coefficient of yield. BMC Plant Biol. 2020;20:1–15. https://doi.org/10.1186/S12870-020-02519-9.
Du Y, Zhao Q, Chen L, Yao X, Zhang H, Wu J, et al. Effect of Drought Stress during Soybean R2–R6 Growth Stages on Sucrose Metabolism in Leaf and Seed. Int. J. Mol. Sci. 2020;21. https://doi.org/10.3390/IJMS21020618.
Do Rosário Rosa V, Da Silva AA, Brito DS, Júnior JDP, Silva CO, Dal-Bianco M, et al. Drought stress during the reproductive stage of two soybean lines. Pesqui. Agropecuária Bras. 2020;55: 1–11. https://doi.org/10.1590/S1678-3921.PAB2020.V55.01736.
Huang X, Han B. Natural variations and genome-wide association studies in crop plants. Annu Rev Plant Biol. 2014;65:531–51. https://pubmed.ncbi.nlm.nih.gov/24274033/.
Dhanapal AP, Ray JD, Singh SK, Hoyos-Villegas V, Smith JR, Purcell LC, et al. Genome-wide association mapping of soybean chlorophyll traits based on canopy spectral reflectance and leaf extracts. BMC Plant Biol. 2016;16(1):1–15. https://doi.org/10.1186/s12870-016-0861-x.
Wang L, Yang Y, Zhang S, Che Z, Yuan W, Yu D. GWAS reveals two novel loci for photosynthesis-related traits in soybean. Mol Genet Genomics. 2020;295(3):705–16. https://doi.org/10.1007/s00438-020-01661-1.
Saleem A, Muylle H, Aper J, Ruttink T, Wang J, Yu D, et al. A Genome-Wide Genetic Diversity Scan Reveals Multiple Signatures of Selection in a European Soybean Collection Compared to Chinese Collections of Wild and Cultivated Soybean Accessions. Front Plant Sci. 2021;12:256. https://doi.org/10.3389/fpls.2021.631767.
PLAZA 4.0: an integrative resource for functional, evolutionary and comparative plant genomics. [accessed 2021 Oct 31]. https://bioinformatics.psb.ugent.be/plaza/versions/plaza_v4_dicots/.
The SoyBase Database. [accessed 2021 Oct 31]. https://www.soybase.org/.
Khrouchtchova A, Hansson M, Paakkarinen V, Vainonen JP, Zhang S, Jensen PE, et al. A previously found thylakoid membrane protein of 14 kDa (TMP14) is a novel subunit of plant photosystem I and is designated PSI-P. FEBS Lett. 2005;579(21):4808–12. https://doi.org/10.1016/j.febslet.2005.07.061.
Bang WY, Jeong IS, Kim DW, Im CH, Ji C, Hwang SM, et al. Role of Arabidopsis CHL27 Protein for Photosynthesis, Chloroplast Development and Gene Expression Profiling. Plant Cell Physiol. 2008;49(9):1350–63. https://academic.oup.com/pcp/article/49/9/1350/1885468.
Liu J, Hua W, Hu Z, Yang H, Zhang L, Li R, et al. Natural variation in ARF18 gene simultaneously affects seed weight and silique length in polyploid rapeseed. Proc Natl Acad Sci U S A. 2015;112(37):E5123–32. https://doi.org/10.1073/pnas.1502160112.
Ikeyama Y, Tasaka M, Fukaki H. RLF, a cytochrome b(5)-like heme/steroid binding domain protein, controls lateral root formation independently of ARF7/19-mediated auxin signaling in Arabidopsis thaliana. Plant J. 2010;62(5):865–75. https://pubmed.ncbi.nlm.nih.gov/20230485/.
Kishor PBK, Hima Kumari P, Sunita MSL, Sreenivasulu N. Role of proline in cell wall synthesis and plant development and its implications in plant ontogeny. Front Plant Sci. 2015;20(6):1–17. https://doi.org/10.3389/FPLS.2015.00544.
Gattolin S, Sorieul M, Frigerio L. Mapping of Tonoplast Intrinsic Proteins in Maturing and Germinating Arabidopsis Seeds Reveals Dual Localization of Embryonic TIPs to the Tonoplast and Plasma Membrane. Mol Plant. 2011;4(1):180–9. http://www.cell.com/article/S1674205214605700/fulltext.
Ishihara S, Takabayashi A, Ido K, Endo T, Ifuku K, Sato F. Distinct functions for the two PsbP-like proteins PPL1 and PPL2 in the chloroplast thylakoid lumen of Arabidopsis. Plant Physiol. 2007;145(3):668–79. https://pubmed.ncbi.nlm.nih.gov/17827269/.
Palmer AJ, Baker A, Muench SP. The varied functions of aluminium-activated malate transporters–much more than aluminium resistance. Biochem Soc Trans. 2016;44(3):856. https://doi.org/10.1042/BST20160027.
Zygadlo A, Robinson C, Scheller HV, Mant A, Jensen PE. The properties of the positively charged loop region in PSI-G are essential for its “spontaneous” insertion into thylakoids and rapid assembly into the photosystem I complex. J Biol Chem. 2006;281(15):10548–54. https://pubmed.ncbi.nlm.nih.gov/16478728/.
Li H, Yang M, Zhao C, Wang Y, Zhang R. Physiological and proteomic analyses revealed the response mechanisms of two different drought-resistant maize varieties. BMC Plant Biol. 2021;21(1):1–15. https://doi.org/10.1186/s12870-021-03295-w.
North HM, Almeida A De, Boutin JP, Frey A, To A, Botran L, et al. The Arabidopsis ABA-deficient mutant aba4 demonstrates that the major route for stress-induced ABA accumulation is via neoxanthin isomers. Plant J. 2007;50(5):810–24. https://pubmed.ncbi.nlm.nih.gov/17470058/.
Singh S, Yadav S, Singh A, Mahima M, Singh A, Gautam V, et al. Auxin signaling modulates LATERAL ROOT PRIMORDIUM1 (LRP1) expression during lateral root development in Arabidopsis. Plant J. 2020;101(1):87–100. https://pubmed.ncbi.nlm.nih.gov/31483536/.
Xu J, Xing XJ, Tian YS, Peng RH, Xue Y, Zhao W, et al. Transgenic Arabidopsis Plants Expressing Tomato Glutathione S-Transferase Showed Enhanced Resistance to Salt and Drought Stress. PLoS One. 2015;10(9):e0136960. https://doi.org/10.1371/journal.pone.0136960.
Kilili KG, Atanassova N, Vardanyan A, Clatot N, Al-Sabarna K, Kanellopoulos PN, et al. Differential Roles of Tau Class Glutathione S-Transferases in Oxidative Stress *. J Biol Chem. 2004;279(23):24540–51. http://www.jbc.org/article/S0021925820665917/fulltext.
Zou JJ, Li XD, Ratnasekera D, Wang C, Liu WX, Song LF, et al. Arabidopsis CALCIUM-DEPENDENT PROTEIN KINASE8 and CATALASE3 Function in Abscisic Acid-Mediated Signaling and H2O2 Homeostasis in Stomatal Guard Cells under Drought Stress. Plant Cell. 2015;27(5):1445–60. https://academic.oup.com/plcell/article/27/5/1445/6096493.
Fendrych M, Van Hautegem T, Van Durme M, Olvera-Carrillo Y, Huysmans M, Karimi M, et al. Programmed cell death controlled by ANAC033/SOMBRERO determines root cap organ size in Arabidopsis. Curr Biol. 2014;24(9):931–40. https://pubmed.ncbi.nlm.nih.gov/24726156/.
Willemsen V, Bauch M, Bennett T, Campilho A, Wolkenfelt H, Xu J, et al. The NAC Domain Transcription Factors FEZ and SOMBRERO Control the Orientation of Cell Division Plane in Arabidopsis Root Stem Cells. Dev Cell. 2008;15(6):913–22. http://www.cell.com/article/S1534580708004012/fulltext.
Koops P, Pelser S, Ignatz M, Klose C, Marrocco-Selden K, Kretsch T. EDL3 is an F-box protein involved in the regulation of abscisic acid signalling in Arabidopsis thaliana. J Exp Bot. 2011;62(15):5547–60. https://academic.oup.com/jxb/article/62/15/5547/560013.
Huang K, Peng L, Liu Y, Yao R, Liu Z, Li X, et al. Arabidopsis calcium-dependent protein kinase AtCPK1 plays a positive role in salt/drought-stress response. Biochem Biophys Res Commun. 2018;498(1):92–8. https://pubmed.ncbi.nlm.nih.gov/29196259/.
Salas-Muñoz S, Rodríguez-Hernández AA, Ortega-Amaro MA, Salazar-Badillo FB, Jiménez-Bremont JF. Arabidopsis AtDjA3 null mutant shows increased sensitivity to abscisic acid, salt, and osmotic stress in germination and post-germination stages. Front Plant Sci. 2016;7:220. https://doi.org/10.3389/FPLS.2016.00220.
Nick S, Meurer J, Soll J, Ankele E. Nucleus-Encoded Light-Harvesting Chlorophyll a/b Proteins are Imported Normally into Chlorophyll b-Free Chloroplasts of Arabidopsis. Mol Plant. 2013;6(3):860–71. http://www.cell.com/article/S1674205214600459/fulltext.
Ren Z, Zheng Z, Chinnusamy V, Zhu J, Cui X, Iida K, et al. RAS1, a quantitative trait locus for salt tolerance and ABA sensitivity in Arabidopsis. Proc Natl Acad Sci U S A. 2010;107(12):5669–74. https://doi.org/10.1073/pnas.0910798107.
Finkelstein RR, Lynch TJ. The Arabidopsis Abscisic Acid Response Gene ABI5 Encodes a Basic Leucine Zipper Transcription Factor. Plant Cell. 2000;12(4):599–609. https://academic.oup.com/plcell/article/12/4/599/6008808
Bi C, Ma Y, Wu Z, Yu YT, Liang S, Lu K, et al. Arabidopsis ABI5 plays a role in regulating ROS homeostasis by activating CATALASE 1 transcription in seed germination. Plant Mol Biol. 2017;94(1):197. https://doi.org/10.1007/S11103-017-0603-Y.
Guo M, Liu JH, Ma X, Luo DX, Gong ZH, Lu MH. The plant heat stress transcription factors (HSFS): Structure, regulation, and function in response to abiotic stresses. Front Plant Sci. 2016;7:114. https://doi.org/10.3389/FPLS.2016.00114.
Sakuma Y, Maruyama K, Qin F, Osakabe Y, Shinozaki K, Yamaguchi-Shinozaki K. Dual function of an Arabidopsis transcription factor DREB2A in water-stress-responsive and heat-stress-responsive gene expression. Proc Natl Acad Sci U S A. 2006;103(49):18822–7. https://doi.org/10.1073/pnas.0605639103.
Xu YH, Liu R, Yan L, Liu ZQ, Jiang SC, Shen YY, et al. Light-harvesting chlorophyll a/b-binding proteins are required for stomatal response to abscisic acid in Arabidopsis. J Exp Bot. 2012;63(3):1095–106. https://pubmed.ncbi.nlm.nih.gov/22143917/.
Lopez-Molina L, Mongrand S, Kinoshita N, Chua NH. AFP is a novel negative regulator of ABA signaling that promotes ABI5 protein degradation. Genes Dev. 2003;17(3):410–8. http://genesdev.cshlp.org/content/17/3/410.full.
Yang B, Voiniciuc C, Fu L, Dieluweit S, Klose H, Usadel B. TRM4 is essential for cellulose deposition in Arabidopsis seed mucilage by maintaining cortical microtubule organization and interacting with CESA3. New Phytol. 2019;221(2):881–95. https://doi.org/10.1111/nph.15442
Hussain S, Liu T, Iqbal N, Brestic M, Pang T, Mumtaz M, et al. Effects of lignin, cellulose, hemicellulose, sucrose and monosaccharide carbohydrates on soybean physical stem strength and yield in intercropping. Photochem Photobiol Sci. 2020;19(4):462–72. https://pubs.rsc.org/en/content/articlehtml/2020/pp/c9pp00369j.
Kato Y, Sakamoto W. FtsH protease in the thylakoid membrane: Physiological functions and the regulation of protease activity. Front Plant Sci. 2018;9:855. https://doi.org/10.3389/FPLS.2018.00855.
Queitsch C, Hong SW, Vierling E, Lindquist S. Heat Shock Protein 101 Plays a Crucial Role in Thermotolerance in Arabidopsis. Plant Cell. 2000;12(4):479–92. https://academic.oup.com/plcell/article/12/4/479/6008798.
Zhou YP, Duan J, Fujibe T, Yamamoto KT, Tian CE. AtIQM1, a novel calmodulin-binding protein, is involved in stomatal movement in Arabidopsis. Plant Mol Biol. 2012;79(4–5):333–46. https://doi.org/10.1007/s11103-012-9915-0.
Peng L, Shikanai T. Focus Issue on Plastid Biology: Supercomplex Formation with Photosystem I Is Required for the Stabilization of the Chloroplast NADH Dehydrogenase-Like Complex in Arabidopsis. Plant Physiol. 2011;155(4):1629.
Pantalone VR, Rebetzke GJ, Burton JW, Carter TE. Phenotypic Evaluation of Root Traits in Soybean and Applicability to Plant Breeding. Crop Sci. 1996;36(2):456–9. https://doi.org/10.2135/cropsci1996.0011183X003600020039x.
Tanaka Y, Fujii K, Shiraiwa T. Variability of Leaf Morphology and Stomatal Conductance in Soybean [Glycine max (L.) Merr.] Cultivars. Crop Sci. 2010;50(6):2525–32. https://doi.org/10.2135/cropsci2010.02.0058.
Fletcher AL, Sinclair TR, Allen LH. Transpiration responses to vapor pressure deficit in well watered “slow-wilting” and commercial soybean. Environ Exp Bot. 2007;61(2):145–51. https://doi.org/10.1016/J.ENVEXPBOT.2007.05.004.
Ries LL, Purcell LC, Carter TE, Edwards JT, King CA. Physiological Traits Contributing to Differential Canopy Wilting in Soybean under Drought. Crop Sci. 2012;52(1):272–81. https://doi.org/10.2135/cropsci2011.05.0278.
Blum A. Drought resistance, water-use efficiency, and yield potential—are they compatible, dissonant, or mutually exclusive? Aust J Agric Res. 2005;56(11):1159–68. https://www.publish.csiro.au/ar/AR05069.
Wijewardana C, Alsajri FA, Irby JT, Krutz LJ, Golden BR, Henry WB, et al. Water Deficit Effects on Soybean Root Morphology and Early-Season Vigor. Agron. 2019;9(12):836. https://www.mdpi.com/2073-4395/9/12/836/htm.
Prince S, Murphy M, Mutava R, Zhang Z, Nguyen N, Kim Y, et al. Evaluation of high yielding soybean germplasm under water limitation. J Integr Plant Biol. 2016;58(5):475–91. https://pubmed.ncbi.nlm.nih.gov/26172438/.
Piro G, Leucci MR, Waldron K, Dalessandro G. Exposure to water stress causes changes in the biosynthesis of cell wall polysaccharides in roots of wheat cultivars varying in drought tolerance. Plant Sci. 2003;165(3):559–69. https://doi.org/10.1016/S0168-9452(03)00215-2.
Hudak CM, Patterson RP. Vegetative Growth Analysis of a Drought-Resistant Soybean Plant Introduction. Crop Sci. 1995;35(2):464–71. https://doi.org/10.2135/cropsci1995.0011183X003500020031x.
Ray JD, Dhanapal AP, Singh SK, Hoyos-Villegas V, Smith JR, Purcell LC, et al. Genome-wide association study of ureide concentration in diverse maturity group IV soybean [Glycine max (L.) Merr.] accessions. G3 Genes, Genomes, Genet. 2015;5(11):2391–403. https://doi.org/10.1534/g3.115.021774.
Blum A, Tuberosa R. Dehydration survival of crop plants and its measurement. J Exp Bot. 2018;69(5):975–81. https://pubmed.ncbi.nlm.nih.gov/29325054/.
Farooq M, Hussain M, Wahid A, Siddique KHM. Drought Stress in Plants: An Overview. In: Aroca R, editor. Plant Responses to Drought Stress: From Morphological to Molecular Features. Springer, Berlin, Heidelberg; 2012. p. 1–33. https://doi.org/10.1007/978-3-642-32653-0_1.
Mutava RN, Prince SJK, Syed NH, Song L, Valliyodan B, Chen W, et al. Understanding abiotic stress tolerance mechanisms in soybean: A comparative evaluation of soybean response to drought and flooding stress. Plant Physiol Biochem. 2015;86:109–20. https://doi.org/10.1016/J.PLAPHY.2014.11.010.
Schneider JR, Müller M, Klein VA, Rossato-Grando LG, Barcelos RP, Dalmago GA, et al. Soybean Plant Metabolism under Water Deficit and Xenobiotic and Antioxidant Agent Application. Biol. 2020;9(9):266. https://www.mdpi.com/2079-7737/9/9/266/htm.
Sade N, Del Mar Rubio-Wilhelmi M, Umnajkitikorn K, Blumwald E. Stress-induced senescence and plant tolerance to abiotic stress. J Exp Bot. 2018;69(4):845–53. https://academic.oup.com/jxb/article/69/4/845/4036238.
Zwicke M, Picon-Cochard C, Morvan-Bertrand A, Prud’homme MP, Volaire F. What functional strategies drive drought survival and recovery of perennial species from upland grassland? Ann Bot. 2015;116(6):1001–15. https://pubmed.ncbi.nlm.nih.gov/25851134/.
Albacete A, Cantero-Navarro E, Großkinsky DK, Arias CL, Balibrea ME, Bru R, et al. Ectopic overexpression of the cell wall invertase gene CIN1 leads to dehydration avoidance in tomato. J Exp Bot. 2015;66(3):863–78. https://pubmed.ncbi.nlm.nih.gov/25392479/.
Rivero R, Kojima M, Gepstein A, Sakakibara H, Mittler R, Gepstein S, et al. Delayed leaf senescence induces extreme drought tolerance in a flowering plant. Proc Natl Acad Sci U S A. 2007;104(49):19631–6. https://pubmed.ncbi.nlm.nih.gov/18048328/.
Joshi S, Choukimath A, Isenegger D, Panozzo J, Spangenberg G, Kant S. Improved Wheat Growth and Yield by Delayed Leaf Senescence Using Developmentally Regulated Expression of a Cytokinin Biosynthesis Gene. Front Plant Sci. 2019;10:1285. https://doi.org/10.3389/FPLS.2019.01285.
Brevedan RE, Egli DB. Short Periods of Water Stress during Seed Filling, Leaf Senescence, and Yield of Soybean. Crop Sci. 2003;43(6):2083–8. https://doi.org/10.2135/cropsci2003.2083.
Fourcaud T, Zhang X, Stokes A, Lambers H, Körner C. Plant Growth Modelling and Applications: The Increasing Importance of Plant Architecture in Growth Models. Ann Bot. 2008;101(8):1053–63. https://academic.oup.com/aob/article/101/8/1053/161814.
Fatichi S, Leuzinger S, Körner C. Moving beyond photosynthesis: from carbon source to sink-driven vegetation modeling. New Phytol. 2014;201(4):1086–95. https://doi.org/10.1111/nph.12614.
Garcia ME, Lynch T, Peeters J, Snowden C, Finkelstein R. A small plant-specific protein family of ABI five binding proteins (AFPs) regulates stress response in germinating Arabidopsis seeds and seedlings. Plant Mol Biol. 2008;67(6):643–58. https://pubmed.ncbi.nlm.nih.gov/18484180/.
Zhao KX, Chu SS, Zhang XD, Wang LP, Rono JK, Yang ZM. AtWRKY21 negatively regulates tolerance to osmotic stress in Arabidopsis. Environ Exp Bot. 2020;169: 103920. https://doi.org/10.1016/J.ENVEXPBOT.2019.103920.
Peng L, Fukao Y, Fujiwara M, Takami T, Shikanai T. Efficient operation of NAD(P)H dehydrogenase requires supercomplex formation with photosystem I via minor LHCI in Arabidopsis. Plant Cell. 2009;21(11):3623–40. https://pubmed.ncbi.nlm.nih.gov/19903870/.
Borra-Serrano I, De Swaef T, Quataert P, Aper J, Saleem, A, et al. Closing the Phenotyping Gap: High Resolution UAV Time Series for Soybean Growth Analysis Provides Objective Data from Field Trials. Remote Sens. 2020;2. https://doi.org/10.3390/rs12101644
Tukey JW, Exploratory data analysis. Reading. PA: Addison-Wesley; 1977.
Bates D, Mächler M, Bolker B, and Walker S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015;67: 1–48. https://doi.org/10.18637/JSS.V067.I01.
Akaike H. A New Look at the Statistical Model Identification. IEEE Trans Automat Contr. 1974;19:716–23. https://doi.org/10.1109/TAC.1974.1100705.
Araghi GS, Assad MT. Evaluation of four screening techniques for drought resistance and their relationship to yield reduction ratio in wheat. Euphytica. 1998;103:293–9.
Wang J, Chu S, Zhang H, Zhu Y, Cheng H, Yu D. Development and application of a novel genome-wide SNP array reveals domestication history in soybean. Sci Rep. 2016;6(20728):1–10. https://doi.org/10.1038/srep20728.
Huang M, Liu X, Zhou Y, Summers RM, Zhang Z. BLINK: a package for the next level of genome-wide association studies with both individuals and markers in the millions. Gigascience. 2019;8(2):1–12. https://academic.oup.com/gigascience/article/8/2/giy154/5238723.
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, et al. GAPIT: genome association and prediction integrated tool. Bioinformatics. 2012;28(18):2397–9. https://pubmed.ncbi.nlm.nih.gov/22796960/.
Team R core. RStudio: Integrated Development for R. RStudio, PBC, Boston, MA; 2019. http://www.rstudio.com/.
The Phytozome Database. [accessed 2021 Oct 31]. https://phytozome-next.jgi.doe.gov/.
Acknowledgements
The Authors would like to thank the partners of the EUCLEG project, and in particular Marie Pégard from INRAE, for fruitful discussions on GWAS methodologies.
Funding
This project has received funding from the European Union’s Horizon 2020 Program for Research & Innovation under grant agreement no. 727312 (project: “EUCLEG – Breeding forage and grain legumes to increase EU’s and China’s protein self-sufficiency”).
Author information
Authors and Affiliations
Contributions
AS, HM, and IR-R conceived the study. AS and HM performed the analysis. AS and IR-R drafted the manuscript. AS, HM, JA, and IR-R interpreted the results and elaborated the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study complies with relevant institutional, national, and international guidelines and legislation for plant ethics.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Saleem, A., Roldán-Ruiz, I., Aper, J. et al. Genetic control of tolerance to drought stress in soybean. BMC Plant Biol 22, 615 (2022). https://doi.org/10.1186/s12870-022-03996-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03996-w