
Abstract
Nearly 60 – 80 % of intron-containing plant genes undergo alternative splicing in response to either stress or plant developmental cues. RNA splicing is performed by a large ribonucleoprotein complex called the spliceosome in conjunction with associated subunits such as serine arginine (SR) proteins, all of which undergo extensive phosphorylation. In plants, there are three main protein kinase families suggested to phosphorylate core spliceosome subunits and related splicing factors based on orthology to human splicing-related kinases: the SERINE/ARGININE PROTEIN KINASES (SRPK), ARABIDOPSIS FUS3 COMPLEMENT (AFC), and Pre-mRNA PROCESSING FACTOR 4 (PRP4K) protein kinases. To better define the conservation and role(s) of these kinases in plants, we performed a genome-scale analysis of the three families across photosynthetic eukaryotes, followed by extensive transcriptomic and bioinformatic analysis of all Arabidopsis thaliana SRPK, AFC, and PRP4K protein kinases to elucidate their biological functions. Unexpectedly, this revealed the existence of SRPK and AFC phylogenetic groups with distinct promoter elements and patterns of transcriptional response to abiotic stress, while PRP4Ks possess no phylogenetic sub-divisions, suggestive of functional redundancy. We also reveal splicing-related kinase families are both diel and photoperiod regulated, implicating different orthologs as discrete time-of-day RNA splicing regulators. This foundational work establishes a number of new hypotheses regarding how reversible spliceosome phosphorylation contributes to both diel plant cell regulation and abiotic stress adaptation in plants.
Similar content being viewed by others


Introduction
Alternative splicing (AS) is a fundamental process involved in diversifying the cellular repertoire of mRNA transcripts in order to post-transcriptionally regulate gene expression [1]. It can alter the exon composition of an mRNA transcript to produce multiple transcript isoforms per gene; each with the possibility of being translated into unique proteoforms with specific biological functions, activities, or subcellular localizations [2]. Alternatively, it can involve full or partial intron retainment—a phenomenon commonly found in plants [3, 4], which can result in premature termination codons, producing aberrant transcripts that are targeted by non-sense mediated decay (NMD) [5].
In humans, nearly 95 % of all intron containing genes undergo AS [6], while in the plant model organism—Arabidopsis thaliana (At)—recent estimates suggest between 60 – 80 % intron-containing genes are alternatively spliced [7,8,9]. In humans (Hs), mis-regulation of AS has been associated with 15 % of genetic diseases [10], while in plants, several studies have demonstrated the biological importance of AS in plant metabolism [11] and plant development [12,13,14], including seed germination [15] and flowering [16, 17]. AS in plants has also been reported to occur in response to abiotic stress such as drought [18], heat [19], salt [20], and under abscisic acid (ABA) signaling [21, 22], in addition to having broad ranging circadian plant cell regulation implications [23, 24]. Collectively, studies implicate RNA splicing as a critical survival mechanism under abiotic stress and correspondingly, a means by which to potentially enhance climate resilience and other important agronomic traits in crops.
AS is performed by a large nuclear-localized, multi-subunit protein complex called the spliceosome [25]. The highly orchestrated assembly of the spliceosome involves the ordered interaction of small nuclear RNAs (snRNAs) and ribonucleoproteins (RNPs) which together bind to form small nuclear ribonucleoprotein (snRNP) complexes: U1, U2, U4, U5 and U6 [26]. Spliceosome formation and splice site recognition is then facilitated by several splicing factors including members of the SERINE/ARGININE-RICH (SR) proteins [27]. SR proteins are one of the best-characterized non-snRNP proteins in the spliceosome. They are found to possess diverse roles in transcript splicing, such as the recruitment and promotion of snRNP binding to active splicing events, and aiding in splicing catalysis by recognizing and selecting splice sites for constitutive and alternative splicing [28, 29]. SR proteins are defined by a highly conserved N-terminal RNA binding domain and a C-terminal arginine-serine-rich (RS) domain [30], which is the target of reversible phosphorylation [31].
Illustrated by the large number of phosphoproteomic studies and plant protein post translational modification (PTM) repositories reporting extensive phosphorylation of SR proteins and snRNPs [32], PTM viewer [33] https://www.psb.ugent.be/webtools/ptm-viewer/), qPTMplants [34] http://qptmplants.omicsbio.info/, the plant spliceosome and its regulatory proteins (e.g. SR proteins) are a major targets of regulatory phosphorylation events. The phosphorylation and dephosphorylation of RS domains can alter the ability of SR proteins to interact with other splicing related proteins and with RNA, which in turn modifies the pre-mRNA splicing programme [35]. Based on their orthology to known human splicing-related kinases, these phosphorylation events are likely catalyzed by three protein kinase families: the SERINE/ARGININE PROTEIN KINASES (SRPKs), ARABIDOPSIS FUS3 COMPLEMENT (AFC) or Pre-mRNA PROCESSING FACTOR 4 (PRP4Ks) protein kinases.
In humans, the phosphorylation activity and function of the three splicing-related kinase families have been well-established. HsSRPK1 phosphorylates the N-terminal half of the RS domain of SR proteins [36,37,38], while CDC-Like kinase 2 (CLKs), the human AFC orthologs, further phosphorylate SR proteins to generate hyper-phosphorylated SR proteins that then initiate early spliceosome assembly [39]. Alternatively, HsPRP4K phosphorylation activity is required to stabilize snRNP association during spliceosome assembly [40]. However, it is currently unknown if these human splicing-related kinase families parallel in function to those in plants. With plants demonstrating unique functional differences in their AS landscapes compared to metazoans [9], it is likely that the expansion of splicing-related kinase families in plants parallels the need for additional regulation of diverse plant specific processes.
Our understanding of how AS impacts multiple facets of the plant cell environment has begun to be explored at the transcriptional-level, however, how these changes affect translational and post-translational outcomes (e.g. proteoforms) in plants is only in its infancy. For example, to what extent does the phosphorylation activity of splicing-related kinases affect the outcomes of RNA splicing? Therefore, to better understand their conservation across photosynthetic eukaryotes and to elucidate their roles in plant development and stress adaptation, we performed a genome-scale molecular phylogenetic analysis of the three splicing-related kinases families. We then combine this with transcriptomic and bioinformatic analyses of A. thaliana splicing-related kinases to infer their connection to specific biological processes where RNA splicing has been shown to be significantly altered within photosynthetic eukaryotes, specifically diel plant cell regulation and abiotic stress response. Overall, our findings provide a foundational understanding of these potentially important protein kinase families, opening up numerous avenues for future fundamental and applied plant research.
Results and Discussion
Phylogenetic and evolutionary relationships between photosynthetic eukaryote splicing kinases
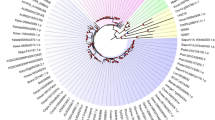
To determine the evolutionary history of the SRPK, AFC, and PRP4K protein families, we constructed phylogenetic trees from a taxonomically diverse set of photosynthetic and non-photosynthetic eukaryotes using a combination of maximum-likelihood and Bayesian analysis approaches (Figs. 1, 2 and 3). Correspondingly, our classification of SRPK, AFC, and PRP4K splicing-related kinases was derived from metazoan orthologs (included here), which are known to impact spliceosome function [40,41,42]. Overall, we see an expansion of each splicing-related kinase family in photosynthetic eukaryotes over evolutionary time, culminating with the emergence of a family organization in land plants that indicates potential diversification of their biological and cellular functions with the colonization of land by plants.

Maximum likelihood phylogenetic tree of SRPK kinases across unicellular and multicellular photosynthetic and select non-photosynthetic organisms. Key nodes are labelled with branch support values from maximum likelihood interference (IQTree), bayesian (Mr. Bayes) and an additional maximum likelihood interference (PhyML), respectively. Node A: (0.97, 1.00, 0.95); Node B (0.98, 0.99, 0.97); Node C: (1.00,1.00, 0.99); Node D: (1.00, 1.00, 1.00); Node E: (1.00, 1.00, 0.80); Node F: (1.00, 0.99, 0.99)

Maximum likelihood phylogenetic tree of AFC kinases across unicellular and multicellular photosynthetic and select non-photosynthetic organisms. Key nodes are labelled with branch support values from maximum likelihood interference (IQTree), bayesian (Mr. Bayes) and an additional maximum likelihood interference (PhyML), respectively. Node A: (1.00, 1.00, 1.00); Node B (1.00, 1.00, 0.99); Node C: (0.97, 1.00, 0.96); Node D: (0.88, 0.93, 0.78); Node E: (0.99, 1.00, 0.99)

Maximum likelihood phylogenetic tree of PRP4K kinases across unicellular and multicellular photosynthetic and select non-photosynthetic organisms. Key nodes are labelled with branch support values from maximum likelihood interference (IQTree), bayesian (Mr. Bayes) and an additional maximum likelihood interference (PhyML), respectively. Node A: (1.00, 1.00, 1.00); Node B: (0.77, 0.78, 0.80); Node C: (0.86, 1.00, 0.90); Node D: (0.98,0.86, 0.95); Node E (0.98, 0.56, 0.96)
SRPK-family
Interestingly, our molecular phylogenetic analysis revealed a discrepancy between the number of protein members in an organism and that organism’s evolutionary position. For example, early unicellular photosynthetic eukaryote Cyanophora paradoxa possesses three SRPK proteins (Fig. 1, Supplemental Table 1), while other organisms appearing later in evolutionary time such as, Volvox carteri have only one SRPK member. This was also noted by Giannakouros and colleagues [43], who found no one-to-one SRPK gene corresponding sequence between evolutionary distant species, including the three SRPK genes of Drosophila melanogaster, which were found to not equate to any one of the three human SRPK genes [44]. These findings suggest independent SRPK duplication events throughout the evolution of distant species resulting in multiple unique SRPK copies in certain taxa. It is possible that this diversification is partially a result of increasing organismal complexity coupled with increased integration of RNA splicing as a regulatory mechanism. Highlighting this hypothesis is HsSRPK1, which is involved in the signal transduction of epidermal growth factors (EGF) through the protein kinase B (Akt) pathway to phosphorylate SR proteins and initiate the downstream pre-mRNA splicing needed for cellular growth [41]. Similarly, Drosophila SRPKs are necessary for the formation of oocyte microtubule spindle assembly and karyosome formation which is critical for proper meiotic division [45].
Despite the disconnect between evolutionary scale and SRPK copy number, there seems to be two SRPK groups that emerge with the evolution of spermatophytes (Fig. 1). Using Arabidopsis as the benchmark, we see AtSRPK1 and AtSRPK2 as part of ‘Group 1’ SRPKs, while AtSRPK3, AtSRPK4, and AtSRPK5 form ‘Group 2’ SRPKs. This suggests that: 1) Group 1 and 2 SRPKs may have arisen through duplication event(s) and 2) there is an evolutionary pressure to maintain two distinct SRPK groups in spermatophytes. Observation of 2 SPRK groups in spermatophytes is consistent with gene duplication and subsequent gene-specific functionalization in land plants, something that has also been associated with whole genome duplication events [46, 47]. However, most duplicated genes are subsequently lost, reverting back to a single gene status [48], while duplicated genes whose function impact core eukaryotic processes are often retained, with proteins involved in signaling and metabolism showing a higher post-duplication retention rate [49]. Thus, the SRPK protein family, which is conserved across eukaryotes and whose function lies at the core of RNA processing regulation, are contenders for functional diversification and selective pressure towards maintaining multiple copies. Further, additional pressure to maintain two SRPK groups may result from evolved differences in substrate specificities and tissue specific expression patterns. For example, human and mice SRPK families have been found to be tissue specific [50, 51], with human SRPKs maintaining tissue specific expression patterns dependent on developmental stage, suggesting functional specialization that parallels organism multicellularity and complexity [52]. Therefore, spermatophytes likely maintained two SRPK groups for their distinct substrate specificities that benefit multicellular functioning.
Alongside our extensive examination of photosynthetic eukaryotes, our study includes select opisthokonts for outgroup comparison. Here, we find the Saccharomyces cerevisiae (budding yeast) SRPK homolog (SRPK1-like Kinase; SKY1) to be most related to those found in photosynthetic eukaryotes, particularly euglenophytes, stramenopiles / heterokonts, alveolates, and rhizaria (SAR) phylogenetic groups (Fig. 1). However, SKY1 contains a glutamate instead of a highly conserved glutamine at position E569 [53]. Interestingly, this residue change may be due to the yeast genome lacking SR protein encoding genes and a near lack of AS in budding yeast [54], implying that SKY1 is a paralog rather than an ortholog of SRPKs. Similarly, the yeast AFC homolog does not group with opisthokonts, advancing the hypothesis that these splicing-related kinases may have different functions in yeast (Fig. 2). Interestingly, our phylogenetic search did not yield a yeast PRP4K ortholog (Fig. 3), however, many of the organisms selected for our phylogenetic search did not have PRP4K ortholog, including rhodophytes with only a few exceptions (Supplemental Figure 1).
AFC-family
Here we found that AFC kinases form three distinct phylogenetic groups (Fig. 2). Using Arabidopsis AFCs as representative proteins, we find AtAFC1 and AtAFC2 to divide into separate groups at the level of monocots and eudicots, while AtAFC3 diverges earlier at the gymnosperm level, suggesting that Group 3 AFCs may be more basal in spermatophytes. Given the formation of these distinct AFC groups across the diverse photosynthetic eukaryotes sampled here, it is probable that AFCs possess specific functions in plants, with AFC1 and AFC2 likely performing cellular and/or biological functions unique to angiosperms.
PRP4K-family
Curiously, unlike the SRPKs and AFCs, we do not find the PRP4Ks to separate into distinct phylogenetic groups, rather clustering at the Brassicaceae family level (i.e. Arabidopsis and Brassica rapa), indicative of more recent gene duplication events. We find this phenomenon as far back as liverworts and other early land plants, while green algae and other photosynthetic secondary endosymbionts (e.g. Emiliania huxleyi) possess only a single PRP4K protein. This suggests that with migration to land, PRP4Ks in photosynthetic eukaryotes have undergone gene duplication events that have seemingly not resulted in orthologs with specialized cellular or biological functions. Alternatively, this may indicate that PRP4Ks perform critical regulatory functions related to RNA splicing in land plants that requires a certain level of genetic redundancy. Further, a third PRP4K (PRP4Kc) seems unique to Arabidopsis since Brassica rapa, a close relative to Arabidopsis, possesses only two PRP4K proteins (Supplemental Table 1), suggesting that PRP4Ks may have been further duplicated in Arabidopsis.
Conservation of protein domain composition across splicing-related kinases
Next, to define gene-function relationships, we endeavored to better understand if protein domain organization parallels our phylogenetic relationships, as it has been found that ortholog identification can be clouded by insertion, deletion, and shuffling of domain architecture [55, 56]. To assess this, we performed a comprehensive domain analysis of the three splicing-related kinase families using a new domain meta-analysis tool called DomainViz [57]. By using DomainViz we were able to deduce the positionality and conservation of protein domains in each splicing-related kinase family.
SRPK-family
Here we uncovered a prominent spacer region located midway through the peptide sequence (Fig. 4). This spacer region bifurcates the kinase domain into two halves creating a bipartite kinase domain, and is specific to the SRPK family. The bipartite kinase domain is present across both unicellular and multicellular eukaryotic organisms suggesting that the evolution of the spacer domain occurred early in the formation of the SRPK protein kinase family and is likely important for protein function. In mammals, the SRPK spacer region functions in localizing the protein to the cytoplasm, and removal of the spacer domain increases SRPK translocation to the nucleus, resulting in the hyper-phosphorylation of SR proteins and initiation of splicing reactions [58]. More precisely, the spacer domain functions as a docking motif by anchoring the SRPK protein to the Hsp70/Hsp90 machinery and thereby restricting SRPK to the cytoplasm [59]. More recent findings suggest that the spacer domain promotes appropriate activation loop folding thus bridging the two catalytic kinase domains into proximity and facilitating the formation of an active conformation [60,61,62]. Interestingly, we found that monocot SRPKs largely lack this spacer region (Fig. 4). Based on linker deletion studies in mammals, this suggests that a sub-population of monocot SRPKs possess a more predominant nuclear localization relative to other photosynthetic eukaryotes and therefore a potentially stronger impact on RNA splicing outcomes.

Comparative phylogenetic analysis of conserved domain across phylogenetics groups: opisthokonts, bryophytes, gymnosperms, monocots, and eudicots. Positionality and length of domain is displayed on the x-axis, while the y-axis represents number of organisms whose peptide sequence contains the identified domain. Domain prediction was acquired through PFAM using DomainViz (http://uhriglabdev.cirrus.ualberta.ca/domainviz; [57])
AFC-family
Unlike SRPKs, the AFC kinase domain is present as a singular unit (Fig. 4). In the phylogenetic groups of monocots, eudicots, and opisthokonts we observed the introduction of a polypeptide extension at the N-terminus with an unspecified function which gives the appearance of a kinase domain shift towards the C-terminus. Interestingly, a small subset of hacrobia AFCs (approximately 10 %) uniquely possesses a RIO1 domain (PF01163) situated in the middle of the protein sequence (Supplemental Figure 1). RIO1 domains are derived from a family of serine kinase domains found in archaea, bacteria, and eukaryotes [63]. In S. cerevisiae, RIO1 is vital for proper cell cycle progression and processing ribosomal RNA [64], which indicates an uniquely adapted function for these AFCs in hacrobia organisms. Similarly, approximately 15 % of chlorophyte protein sequences contain a sulfotransferase-1 domain (PF00685) along with a small percentage of bryophytes possessing a Kdo domain (PF06293) (Fig. 4, Supplemental Figure 1). Both sulfotransferase-1 and Kdo domains have structural similarity to kinase domains [65], which likely highlights some unique structural elements to the AFC kinase domains of chlorophytes and bryophyte AFCs, respectively.
PRP4K-family
Paralleling AFC kinases, the kinase domain of PRP4Ks appears to shift towards the C-terminus in land plants due to the presence of an undefined peptide region in the N-terminal region of the peptide sequence (Fig. 4). We also observe a substantial increase in overall PRP4K protein length along the same evolutionary axis, with eudicots and opisthokonts possessing notably longer PRP4K proteins relative to earlier photosynthetic eukaryotes. Interestingly, almost 20 % of PRP4K proteins in bryophytes contain an atypical protein kinase domain called ABC1 located towards their N-terminus. Similar to the AFCs, this likely indicates additional structural complexity in the kinase domains of PRP4Ks. Proteins maintaining similar ABC1 domains in yeast [66] and Escherichia coli [67] were found to be dually localized to the nucleus and mitochondria, suggesting that some bryophyte PRP4Ks may have unique and unconventional biological functions. Overall, the high degree of domain conservation among phylogenies of the splicing-related kinase families in photosynthetic eukaryotes supports our molecular phylogenetic conclusions and that splicing-related kinase domain conservation is maintained across the domains of life.
Cis-regulatory element motifs of the splicing-related kinases
With protein domain architecture largely conserved within each splicing-related kinase family, we next aimed to define biological and cellular function relationships through a bioinformatic analysis of the cis-regulatory element (CREs) composition of each splicing-related kinase family. Given the lack of known functions for splicing-related kinases in plants, examination of CREs was logical for elucidating functional differences within, and between, families of splicing-related kinases, since CREs play a major role in regulating gene expression. Due to the lack of CRE information across species, we utilized the Arabidopsis SRPK, AFC, and PRP4K kinases as representative genes for this analysis. To extract CREs for the Arabidopsis splicing-related kinase genes, we used AtCisDB [68], which contains predicted and experimentally derived CREs present in the deduced promoter regions of Arabidopsis gene sequences. With Arabidopsis representing the best characterized plant system to date, it represents a reliable proxy for relating CRE information to gene / protein function(s).
Here, we find a collection of established CREs that are SRPK group-specific and group non-specific (Fig. 5). This includes meristematic growth related CREs, such as TELO-box, BELLRINGER (BLR), LFY, and L1-box across the SRPKs, suggesting a critical role in plant development (Fig. 5). We suspect that there may be substrate specificity differences between Group 1 and 2 SRPKs, that when coupled with CRE commonalities, offers a means by which to deploy the SRPK complement needed to initiate particular splicing patterns.

Identification of putative cis regulatory elements (CREs) on the gene sequence of SRPKs, AFCs, and PRP4Ks. Presence of CREs is denoted by dark blue while absence is denoted by light blue. Data was acquired by mining AtCisDB database (https://agris-knowledgebase.org/AtcisDB/; [68])
Despite being divergent phylogenetically, members of the AFC family share a common subset of CREs that include: T-box, W-box, LFY, and RAV1-A (Fig. 5). The presence of a W-box in all AFC promoter sequences indicates the core involvement of all AFC family members in pathogen attack and abiotic stress response. The W-box promoter sequence is responsible for the expression of pathogen defense related genes across multiple plant species [69,70,71], while more recent descriptions of the W-box CRE demonstrated a role in the down-regulation of genes induced by heat and salinity stress [72]. Of the AFCs, AFC2 possesses a comparatively large number of CREs, suggesting that AFC2 may performing the majority of AFC-mediated spliceosome regulation in land plants. Overall splicing-related protein kinases share promoter sequences predominantly involved in developmental and abiotic stress pathways.
Transcriptional expression patterns of the splicing-related kinases indicate diversification of biological and cellular responses
In plants, RNA splicing has been investigated for its role in a number of biological and cellular processes. This has included: abiotic stress responses [9, 73, 74], development [75, 76], and diel plant cell regulation [77]. These disparate studies have involved examining cold [78], heat [79, 80], osmotic [20] stress responses, along with developmental traits such as flowering [81] and the circadian clock [82]. Each of these studies has revealed RNA splicing to be a central element in plant cell regulation and overall plant biology. Correspondingly, we next mined well-established, publicly available gene expression datasets, such as Genevestigator ([83], https://genevestigator.com/) and ePlant ([84], https://bar.utoronto.ca/eplant/), in addition to performing NanoString transcriptomic analysis of all Arabidopsis splicing-related kinases to elucidate their gene expression dynamics in response to plant development, abiotic stress response, and diel plant cell regulation.
Developmental expression
From our CRE analysis, we found specific splicing-related kinases within each family to possess promoter sequences related to plant development, such as BLR, LFY, and RAV1-A promoter sequences. These CREs are associated with genes involved in a multitude of developmental processes such as flowering (Fig. 5), with flowering impacted by the AS of CIRCADIAN CLOCK ASSOCIATED 1 (CCA1), a core circadian clock gene [85]. Mammalian SRPK family members exhibit tissue-specific expression profiles characteristic of specialized functions required for multicellular development [50, 86]. Correspondingly, it is possible that the functional diversification of the plant splicing-related kinases is driven by their tissue / organ expression profiles that differ according to developmental time. Therefore, in order to dissect the roles of these splicing-related kinases in plant development, we analyzed transcript expression levels of the Arabidopsis splicing-related kinases at major developmental stages using gene expression data from ePlant and Genevestigator.
Cell differentiation & organ development
We find numerous developmental related CREs across the Arabidopsis SRPK, AFC, and PRP4K families (Fig. 5). For example, the TELO-box CRE, present in the promoter regions of AtSRPK1, AtSRPK4, and AtSRPK5, is associated with translation-related genes in root meristems such as eukaryotic elongation factor 1 alpha (eEF1A) and several ribosomal protein genes [87,88,89]. eEF1A is expressed in the dividing cells of the root primordia to assist with cytoskeleton formation in germinating seeds, embryos, shoot and root meristems [90]. AtSRPK1 (Log2 seedling root expression = 6.39, Log2 mature root expression = 6.47), AtSRPK2 (Log2 seedling root expression = 6.48, Log2 mature root expression = 6.53), and AtSRPK5 (Log2 seedling root expression = 6.33, Log2 mature root expression = 6.28) are all highly expressed in the roots (Fig. 6), implicating these AtSRPKs in cell expansion. Alternatively, AtSRPK3 is highly expressed during seedling germination (Log2 = 7.32), indicating expression in the early stages of organ growth and development. Similarly, the L1-box, present only in the AtSRPK5 promoter, is associated with genes controlling the growth and development of the outermost layer (L1) of the shoot apical meristem [91]. We also find AtSRPK2, AtSRPK3, AtSRPK4, and AtSRPK5 to increase in expression at the shoot apex from the vegetative to inflorescence stage, suggesting that these SRPKs may be involved in cell differentiation (Fig. 6; Supplemental Table 2). The potential involvement of AtSRPKs in cell differentiation and organ development coincides with crucial roles for HsSRPKs in neurodevelopment [92] and in catalyzing the life-beginning event of parental genome reprogramming in the fertilized oocyte [93].

Relative transcript abundance at various stages of Arabidopsis development, from seed to senescence. Values were acquired from BAR ePlant and absolute values were log2 transformed (https://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi; [94])
All members of the AtAFC and AtPRP4K families have a RAV1-A promoter which is involved in the development of rosette leaves and lateral roots (Fig. 5) [95]. AtPRP4Ks demonstrate increased expression in the young rosette (Log2 _AtPRP4Ka = 6.38, Log2 _AtPRP4Kb/c = 6.67), vegetative rosette (Log2 _PRP4Ka = 6.48, Log2 _AtPRP4Kb/c = 6.95), seedling (Log2 _AtPRP4Ka = 6.69, Log2 _AtPRP4Kb/c = 6.90) and mature root (Log2 _AtPRP4Ka = 6.52, Log2 _AtPRP4Kb/c = 6.89) developmental stages (Fig. 6). AtAFC2 also shows expression increase during young rosette (Log2 = 7.41), vegetative rosette (Log2 = 7.23), seedling (Log2 = 7.36) and mature root (Log2 = 7.40) developmental stages (Fig. 6). Interestingly, HsCLKs, the AFC human orthologs, phosphorylate SR splicing factors necessary for the Wnt pathway in human mesenchymal stem cells, which plays a central role in organogenesis, cell differentiation, and tissue remodeling [96, 97]. CLKs have also been implicated in the AS regulation of HMGA2, a gene required for human hematopoietic stem cell development [98]. Correspondingly, this positions AFC splicing-related kinases as critically important developmental regulators.
Flowering & seed maturation
We find several flowering related CREs in the promoters of AtSRPK genes (Figs. 5 and 6). BLR (present in the AtSRPK5 promoter) and LFY (present in the AtSRPK1, AtSRPK4, and AtSRPK5 promoters) in particular, are associated with floral genes that drive floral organ development [99, 100]. Correspondingly, these CREs align with high AtSRPK gene expression in reproductive developmental stages beginning at the shoot apex inflorescence through to dry seed (Fig. 6, Supplemental Figure 2, Supplemental Tables 2 and 3). On a whole, AtSRPKs have reduced expression in mature flower, with only a marginal increase at bolting (Supplemental Figure 2, Supplemental Table 3). However, when we contrast this with their expression levels within individual floral organs, we find exceptionally high, organ-specific AtSRPK1 and AtSRPK2 expression in the stamen (Log2 _AtSRPK1 = 6.77, Log2_AtSRPK2 = 6.96) and pollen (Log2 _AtSRPK1 = 9.34, Log2 _AtSRPK2 = 9.60), suggesting that SRPKs may be required for the initiation of reproductive related AS events.
PRP4Ks may also be involved in developmental processes due to the presence of auxin response factor CREs (ARF and ARF1) present in the AtPRP4Kc promoter. ARFs have been implicated in development through loss-of-function mutant analysis [101,102,103], with ARF1 controlling leaf senescence and floral organ abscission in Arabidopsis [104]. Interestingly, AtAFC2, AtAFC3, and AtPRP4Ks also have high expression in the male associated flower organs (Fig. 6, Supplemental Table 2). Further, Kanno and colleagues (2018) found that prp4ka mutant plants possess delayed flowering, implicating PRP4Ka in flower organ development. The same group reported that the prp4ka prp4kb double mutant was not viable, emphasizing their integral involvement in plant reproduction.
Beyond organ flower development, it seems select splicing-related kinases (AtSRPK1, AtSRPK4, AtAFC2, AtAFC3, and AtPRP4Ka) may be involved in silique maturation. Expression levels increase steadily throughout silique maturation culminating with peak expression levels in dry seeds (Fig. 6). AtSRPK1 is particularly elevated at all stages of silique maturation and has the highest expression level in dry seeds (Supplemental Table 2). Similarly, AtAFC2, AtAFC3, and AtPRP4Kb/c each have considerable increases in dry seed expression (Log2 = 8.59, Log2 = 8.17, Log2 = 7.39, Log2 = 7.39, respectively) relative to other tissues (Fig. 6). Interestingly, recent transcriptome profiling of dry seeds found that while overall transcription declined in dry seeds, AS increased [105]. This is specifically highlighted by AS regulation of PHYTOCHROME INTERACTING FACTOR 6 (PIF6), whose AS variant demonstrates reduced seed dormancy [106].
Senescence
Following silique maturation, we find select splicing-related kinases (AtSRPK1, AtSRPK4, AtAFC2, AtAFC3, and AtPRP4Kb/c) to possess increased expression at senescence (Fig. 6, Supplemental Figure 2, Supplemental Tables 2 and 3). In particular, AtSRPK4, AtAFC2 and AtPRP4Kb/c sharply rise in their expression at senescence. In poplar trees (Populus tomentosa) a splice variant of the NAC transcription factor PtPD26 was found to regulate numerous other NAC transcription factors which delay leaf senescence [107]. We suspect that these splicing-related kinases play a role in cell cycle progression or cell death through the phosphorylation of splicing factors required for the splicing of senescence related genes. Currently, our understanding of the extent to which AS plays a role in development remains largely unresolved, however, our data indicates that future experimentation should focus on elucidating the role these specific splicing-related kinases play in modulating plant death.
Abiotic stress
Abiotic stresses such as drought, salt, heat, and cold, demand accurate and rapid transcriptional modulation for successful adaptation by plants. Transcriptomic studies examining these stresses have found extensive global transcriptome changes in response to osmotic, salt, heat, and cold stresses that occur within minutes to hours of induced stress. For example, 42 %, 46 %, and 53 % of the Arabidopsis transcriptome had a greater than two-fold change from 3 to 27 h of 4 ºC, 200 mM mannitol, and 100 mM NaCl stress, respectively [108]. Previous work by Calixto and colleagues (2018) reported rapid global transcriptional change and AS in response to cold stress of which many were splicing factors and other RNA binding proteins. Such rapid transcriptional change is likely to include fluctuating activities of upstream regulators such as splicing-related kinase. Hence, we sought to investigate the potential involvement of splicing-related kinases in abiotic stress response using ePlant and Genevestigator databases, in addition to acquiring new transcriptome data as part of this study. To do this, we quantified the expression of all 11 splicing-related kinases in either the shoots or roots under osmotic, salt, heat, and cold stress using experimental conditions that parallel those tested in Kilian et al., 2007. Correspondingly, we subjected Arabidopsis seedlings to 300 mM mannitol to simulate osmotic stress and 150 mM NaCl for salt stress, while heat stress involved a 38 ºC exposure for 3 h followed by a 3 h recovery, which is the time at which the majority of splicing-related kinase genes experienced the highest transcriptional changes (Supplemental Figures 3, 4 and 5, and Supplemental Table 4). For cold-stress treatments, Arabidopsis seedlings were exposed to 4 ºC for 24 h.
Osmotic & salt stress
Both AtSRPK1 and AtSRPK5 possess CREs indicating involvement in drought-related response pathways due to their shared DPBF1&2, MYB4, and ATB2 promoter sequences, all of which have been shown to regulate the expression of genes related to drought and in the abscisic acid (ABA) mediated response pathways [109, 110]. Correspondingly, AtSRPK1 exhibited a significant increase in expression under both salt and osmotic stress (Fig. 7). AtSRPK3 also maintained a significant increase in root (Log2FC = 1.26, q-value ≤ 0.02) and shoot (Log2FC = 1.88, q-value ≤ 0.01) expression under salt stress (Fig. 7, Supplemental Figure 3, Supplemental Tables 4 and 5). We also see a significant increase in AtSRPK4 expression in shoots (Log2FC = 1.44, q-value ≤ 0.03) upon osmotic stress, while AtSRPK2 is the only AtSRPK that significantly decreases in shoot expression under osmotic stress (Log2FC = -1.37, q-value ≤ 0.02). Interestingly, despite AtSRPK5 possessing a number of drought-related promoter sequences (Fig. 5), we found no significant change in its expression relating to osmotic and salt stress, suggesting that AtSRPK5 induction in response to drought may occur at specific stages of plant development or in specific organs not sampled here.

Relative Log2 fold change of splicing-related kinases transcript abundance under abiotic stresses: osmotic, salt, heat, and cold stress. Each stress was induced with parallel parameters from Kilian et al., 2007. 4 replicates were averaged and the comparison to control were FDR adjusted p-values
Both the AtAFC and AtPRP4K families also possess a handful of ABA-related CREs (Fig. 5). For example, the promoters of AtAFC1 and AtAFC2 have an ABA-responsive element (ABRE-like) CRE, which is involved in osmotic stress response [111], while AtAFC2 and AtAFC3 both maintain a Dc3 promoter-binding factor (DPBF1&2) CRE, which is ABA and water stress response related [109, 112]. Despite the presence of these drought-stress related CREs, we find that AtAFCs generally decrease in expression under salt conditions and increase their expression under osmotic conditions (Fig. 7). In particular, AtAFC1 significantly decreases (Log2FC = -1.38, q-value ≤ 0.01) under salt stress in the roots, while AtAFC3 significantly decreases (Log2FC = -1.57, q-value ≤ 0.001) under osmotic stress in the shoots. AtPRP4Ks also have drought-stress related CREs, such as RAV1-A, DPBF1&2, and W-box (Fig. 5), but were not significantly influenced by any of the stressors applied in our study outside of AtPRP4Kc under salt conditions (Root_Log2FC = 1.83, q-value ≤ 0.05) (Fig. 7, Supplemental Table 6). This finding contradicts the previous hypothesis that AtPRP4Kc is a pseudogene [14], indicating that AtPRP4Kc is an expressed gene that has specific roles in abiotic stress response. It is possible however, that the transcriptional changes in other AtPRP4K genes were not captured by our harvesting strategy as it was end-point based.
Heat stress
Although we did not specifically find heat-related CREs on any of the splicing-related kinases (Fig. 5), we do see a significant impact of heat on AtSPRK1 expression (Fig. 7). AtSRPK1 uniquely demonstrates a significant increase (Log2FC = 2.1, q-value ≤ 0.001) under heat relative to the other AtSRPKs. This was unexpected, since the ePlant microarray data found AtSRPK3 and AtSRPK4 exhibiting differential expression under heat stress, while AtSRPK1 remained relatively unchanged (Supplemental Figure 3, Supplemental Table 4). Again, our biological sample size provides a more robust depiction of stress induced changes in expression relative to previous studies. We observe AtSRPK1 significantly increasing under the majority of the stresses compared to the other members of the AtSRPK family, suggesting that AtSRPK1 may be specifically required for AS of certain genes central to abiotic stress responses.
HsSRPKs have been classified as “stress kinases” due to their unique position in transmitting cellular stress signals from the cytoplasm to the nucleus through their phosphorylation of SR splicing factors activating their translocation into the nucleus to induce splicing in response to stress signals [60]. For example, sorbitol-induced osmotic stress in mammalian cells resulted in a sufficient osmotic shock to dissociate HsSRPK1 from its chaperone complexes resulting in its translocation from the cytoplasm to the nucleus [113]. Further, upon treatment with paraquat (a compound that induces superoxide formation and oxidative stress) human neuroblastoma cells increase HsSRPK2 nuclear translocation to adjust the splicing pattern of genes involved in DNA repair, cell cycle control, and apoptosis [114]. When combined with the drought expression changes in AtAFC1 and AtAFC3 along with AtPRP4K, our results suggest that splicing-related kinases, in particular SRPKs, have a broad role in mediating plant drought-like responses.
Cold stress
AtSRPK3 and AtSRPK4, both belonging to Group 2 SRPKs, share a dehydration-responsive element (DRE-like) CRE, which is important for the transcriptional regulation of cold-responsive genes [110] and support our hypothesis that there is functional specificity to Group 1 and 2 SRPKs. Although only AtSRPK3 and AtSRPK4 have a cold-related CRE, we find that cold stress induced a significant increase in all AtSRPKs transcript levels across both shoots and roots except for AtSRPK2 in shoots (Fig. 7, Supplemental Figure 3). Lack of AtSRPK2 transcriptional change in response to cold correlates with the overall lack of CREs present in the AtSRPK2 promoter (Fig. 5). It is likely that AtSRPK2 is either constitutively expressed or has CREs not captured by the AtCisDB database. As such, AtSRPK2 is likely a constitutively active Group 1 SRPK member while AtSRPK1 may be a Group 1 stress-responsive SRPK.
Alternatively, AtAFC2 and AtAFC1/3 show opposite shoot expression patterns when exposed to cold treatment. AtAFC2 significantly increases (Log2FC = 1.55, q-value ≤ 0.01), while AtAFC3 significantly decreases (Log2FC = -1.42, q-value ≤ 0.02). Interestingly, The human HsCLK orthologs of plant AFCs, have been shown to be important thermo-sensors that are required for temperature-responsive AS to adjust the circadian biology of mammals [115]. In mammals, lower body temperatures activates CLKs, resulting in increased phosphorylation of SR proteins. Moreover, AFC orthologs in other animal systems are also temperature sensitive, such as in turtle (Trachemys scripta) and fruit fly (D. melanogaster), which show reduced CLK protein activity above their preferred living temperatures that corresponds to an AS change mediated by temperature dependent CLK activity [115]. Since AFCs are highly conserved across photosynthetic eukaryotes, coupled with the dynamic change in expression observed here in response to cold, it is likely that AFCs also have a role in temperature perception and acclimation in planta as well.
AtPRP4K expression patterns were primarily impacted by cold stress (Fig. 7), with both AtPRP4Ka (Log2FC = 1.32, q-value ≤ 0.02) and AtPRP4Kb (Log2FC = 2.61, q-value ≤ 0.01) significantly increasing in their expression in shoots as a result of cold stress. Interestingly, fission yeast PRP4K was discovered as a temperature-sensitive mutant defective in RNA splicing [116, 117], suggesting broader evolutionary roles for PRP4Ks in cold-stress responses. Overall given the lack of divergence amongst orthologous PRP4Ks across land plants, coupled with the lack of CRE differences amongst the AtPRP4Ks, it is likely that PRP4Ks serve largely redundant roles in relation to abiotic stress response.
Light regulated expression
Recently, co-transcriptional regulation, such as AS, has been highlighted as a mechanism by which plants regulate their internal circadian clock [118]. Correspondingly, regulators of splicing, such as SRPKs, AFCs, and PRP4Ks, may then be involved in the timing of gene expression for circadian clock function. Firstly, we found that various members of the Arabidopsis SPRK, AFC, and PRP4K families have light-dependent CREs in their promoter regions (Fig. 5). In particular, AtSRPK1, AtSRPK3, and AtSRPK4 which possess light responsive CREs such as SORLIP2, GATA, and T-box. Alternatively, PRP4Ka has an ATB2 CRE which has been shown to be involved in energy supply and demand, while being regulated by light and hypo-osmolality [119, 120]. As well, all AtPRP4K members also maintain RAV1-A and SORLIP2 CRE sequences. SORLIP2 is responsive to signals transmitted by the phytochrome A photoreceptor pathway [121], which in C. reinhardtii shows a strong light dose-dependent activation [122]. This suggests that AtSRPK1, AtSRPK3, AtSRPK4, and AtPRP4Ks may be light-activated as well. Therefore, to study how the presence of light-dependent CREs translates to gene expression changes, we mined DiurnalDB [123] to study diel and photoperiodic expression levels of all 11 splicing-related kinase genes (Supplemental Figure 6, Supplemental Table 5). We then experimentally quantified how the transcript level of these splicing-related kinases change throughout the day under 8:16, 12:12, 16:8 and 24:0 photoperiod conditions by measuring Zeitgeber time (ZT) 6, 11, 18, and 23.
SRPK-family
Under an 8:16 photoperiod we find that all AtSRPKs have a peak expression at mid-day through to the evening (ZT11, ZT18), while under a 12:12 photoperiod, AtSRPK1 and AtSRPK2 have a peak expression at ZT18 and ZT23 (Fig. 8; Supplemental Table 7). Under long-day photoperiod conditions (16:8), all SRPKs exhibited peak expression at ZT23. Taken together, our SRPK transcript expression data indicates that they may be required for the day-to-night transitions; the same time-point that CCA1 undergoes AS [85]. Furthermore, the increasing photoperiod shift of SRPK peak expression towards the end-of-night (ZT23) suggests that SRPKs may be involved in processes controlled by light signals such as flowering.

Relative transcript abundance under various photoperiods: 8 h: 16 h, 12 h: 12 h, 16 h: 8 h, 24 h: 0 h. Normalized values were log2 transformed and averaged across replicates
AFC-Family
Similarly, we find that AtAFCs maintain peak expression at ZT11 and ZT18 under 8:16 photoperiod (Fig. 8; Supplemental Table 6). The AtAFC family also experiences a peak expression shift towards ZT23 upon lengthening of daylight. Since we found various flowering and developmental CREs in the promoters of AtAFC genes, they may be involved in diel determined flowering control (Fig. 5). Alternatively, the circadian clock has been shown to be tightly intertwined with age-dependent senescence. ORE1, a positive regulator of age induced senescence increases in levels under long day periods and CCA1 directly suppresses ORE1 delaying senescence [124]. Given that high AtAFCs transcript levels are found during leaf senescence (Fig. 6, Supplemental Figure 2), it is possible that AFCs are driven by the circadian clock and required for age-dependent senescence.
PRP4K-family
Lastly, we see that AtPRP4Ks possess a peak expression at mid-day (ZT11, ZT18) under 8:16 photoperiod, with AtPRP4Kc exhibiting a weak diel expression pattern under all photoperiod conditions (Fig. 8). Like the other splicing-related kinase families, AtPRP4Ks experience a shift in expression towards ZT23 as day length increases. Collectively, these findings indicate that splicing-related kinases, as a whole, may be highly regulated by light signals.
The significantly increased expression levels of the majority of the splicing-related kinases under cold treatment combined with their elevated expression at end-of-night time-points suggests that they may be involved in regulating temperature dependent AS (Fig. 8). Patterns of clock gene alternative splicing are influenced by changes in photoperiod and abiotic stresses [85], with evidence that AS is involved in the expression of clock gene splice variants at low temperatures [82]. For example, LHY protein abundance is reduced at low temperatures due to the retention of the first intron in the LHY 5’-UTR, resulting in NMD [125]. While CCA1 AS is suppressed by low temperature resulting in an unidirectional production of the CCA1a (an intron retention splice variant) that induces freezing tolerance [126]. Furthermore, Filichkin and collogues (2015) showed that SR45 splicing factor may be involved in regulating intron retention of CCA1. Initiation of SR45 splicing activity is induced by protein phosphorylation and temperature fluctuations [127]. SR45 phosphorylation may be performed by AFC2, which has been shown to phosphorylate SR45 in vitro [128]. Together, this places AFC-family splicing-related kinases as potential regulators clock gene AS. Given the regulatory dominance of the clock over global transcription and the regulatory role of AS in gene expression, it is conceivable that AS may act to bridge between temperature perception with downstream processes [129]. The extent to which regulatory splicing-related kinases, such as the AtSRPKs, AtAFCs, and AtPRP4Ks, are involved in attenuating AS in response to thermo-sensing and photoperiod signals and how these signals change the clock remains to be explored.
Potential roles for SRPKs beyond RNA splicing
Many of the roles identified for HsSRPKs have been related to RNA splicing of genes involved in developmental and stress response [60, 93, 113, 114]. However, evidence suggests that HsSRPKs have functions beyond RNA splicing [92]. For example, HsSRPK1 has been found to phosphorylate human protamine 1, an arginine-rich protein involved in histone replacement during the development of mature spermatozoa [130]. Additional reports indicate HsSRPKs phosphorylate other non-splicing related, RS-motif containing proteins [43, 131,132,133]. This raises the possibility that AtSRPKs may have phosphorylation targets other than SR splicing factors and that AtSRPKs may be involved in pathways beyond RNA splicing of abiotic stress or developmental-related genes.
As such, we performed a RS-motif search using the Arabidopsis proteome to identify potential non-splicing related proteins that contain canonical RS motif(s) using ScanProsite [92]. A total of 37 RS-motif hits corresponding to a total of 20 proteins were found, with 17 of the 20 identified proteins representing RNA splicing related proteins, and a total of 3 representing non-splicing related proteins (Supplemental Table 9). Of these 3, DEAD-box ATP-dependent RNA helicase 40 (RH40, AT3G06480) is involved in nonsense-mediated mRNA decay and ribosome biogenesis. While Peptidyl-prolyl cis–trans isomerase CYP95 (PPIase CYP95, At4G32420) accelerates folding of proteins by catalyzing the cis–trans isomerization of proline peptide bonds. Both, RH40 and CYP95, are involved in the post-transcriptional processing of mRNA, suggesting that AtSRPKs may be involved in mRNA degradation pathways or mRNA translation.
No consensus RS motif has been established in Arabidopsis, however the aforementioned motif was used since it was used to identify RS motif containing proteins in Mus musculus proteome [92]. Canonical SR splicing factors proteins were identified using the aforementioned motif search suggesting that this motif is sufficient for identifying RS motif containing plant proteins. However, plants possess divergent SR protein sequences [30], as such, the classification of plant SR proteins does not lie in its specific RS tandem repeat sequences but rather a minimum (20 %) of RS or SR dipeptide composition in the RS domain spanning at least 50 amino acids. Therefore, AtSRPKs may have evolved the capacity to phosphorylate varying RS domains. Future studies will be required to identify the breath of RS sequences phosphorylated by AtSRPKs.
Conclusion
Post-transcriptional splicing of pre-mRNA can produce unique transcript isoforms that may be required for stress adaptation or for the timing of specific developmental stages, and thus represent an important means by which the cell can fine-tune gene expression [1]. RNA splicing is performed by the spliceosome, whose activity is directed and mediated by SR splicing factors [25]. Upstream regulators of splicing, such as splicing-related kinases, are capable of transmitting external signals to the spliceosome by phosphorylating SR proteins thereby activating their translocation to the nucleus and the initiation of splicing [39, 41]. To date, very few studies have looked at splicing-related kinases’ ability to modulate splicing factor activity and pre-mRNA splicing in plants. Understanding how splicing-related kinases transmit external cues to the spliceosome could provide key insights into the regulatory splicing programme of the plant cell. In this study, we present the first genome-scale analysis of the major splicing-related kinase families of photosynthetic eukaryotes. We find that these splicing-related kinases may have both developmental and abiotic stress related promoter sequences as well as significant expression pattern changes in response to cold, osmotic, and salt stress. Furthermore, the kinase families experience diel expression patterns and shifts upon photoperiod lengthening, suggesting that these kinases may be tightly controlled by light and circadian cues, offering new connections to the timing of critical developmental stages, such as flowering. How splicing kinases in plants impact AS remains to be resolved, however, based on their roles in humans and our research, future research will likely implicate their involvement numerous abiotic stresses through the regulation of AS.
Materials and methods
Phylogenetic trees
Amino acid sequences of the protein families were acquired from TAIR (https://www.arabidopsis.org/) (SRPK1; AT4G35500, SRPK2; AT2G17530, SRPK3; AT5G22840, SRPK4; AT3G53030, SRPK5; AT3G44850, AFC1; AT3G53570, AFC2; AT4G24740, AFC3; AT4G32660, PRP4Ka; AT3G25840, PRP4Kb; AT1G13350, PRP4Kc; AT3G53640). Amino acid sequences were aligned using MAFFT, version 7 (http://mafft.cbrc.jp/alignment/server/; [134]) and input into HMMER3 v3.3.2 (http://hmmer.org/) to acquire a protein profile. Organism proteomes were acquired from Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html). Protein family profiles were used to query against full proteomes using HMMER3. For organisms whose proteome is not available, their orthologs were acquired by using the 1KP database [135] using BLASTp (https://db.cngb.org/blast/blast/blastp/?project=onekp). Organisms whose orthologs were acquired using full proteomes or using the 1KP project (https://sites.google.com/a/ualberta.ca/onekp/; Supplemental Table 1). Hit above natural e-value cut off range were select for reciprocal BLAST against Arabidopsis proteome to ascertain orthology. Compiled sequences were aligned using MAFFT E-NS-I for SRPK and MAFFT L-INS-I for AFC and PRP4K. Alignment was inspected using TCS (http://tcoffee.crg.cat/apps/tcoffee/do:core) and manually trimmed using GeneDoc (insert software citation) to remove gaps containing higher than 90 % gaps between sequences (Supplemental File 1, 2, and 3). The first maximum likelihood tree was generated using IQtree ([136] http://iqtree.cibiv.univie.ac.at/) using 1000 bootstrap alignments, 1000 iterations, and 0.99 minimum correlation coefficient parameters. The Baseyian tree was generated using Mr.Bayes with CIPRES ([137] https://www.phylo.org/) with the following parameter: 50 000 000 ngen MCMC, nruns = 2, nchains = 4. Maximum likelihood = 10,000 bootstraps, 1000 iterations, min r2 = 0.99 (Supplemental File 5). The second maximum likelihood tree was generated using PhyML version 3.0 (Guindon et al., 2011; http://www.atgc-montpellier.fr/phyml/, [138]) with LG amino acid substitution model, aLRT SH-like fast likelihood-based method for branch support, and all other parameters set as default.
Domain conservation analysis
The putative orthologous amino acid sequences that were found using HMMER (http://hmmer.org/) and 1kp project were compiled for domain analysis (Supplemental Table 1). Compiled peptide sequences were separated into the following phylogenetic groups: opisthokonts, SAR (stramenopiles, alveolates, rhizaria), hacrobia, rhodophytes, chlorophytes, bryophytes, gymnosperms, monocots, and eudicots. Each phylogenetic group was inputted into DomainViz ([57] https://uhrigprotools.biology.ualberta.ca/domainviz) with the following settings: minimum domain prevalence (0.05) and minimum domain position conservation (0.05).
Percent similarity of HsSRPKs and AtSRPKs protein sequences
Human and Arabidopsis SRPK compiled sequences were aligned using MAFFT LINS-I with default settings. Resulting alignment was inputted through Sequence Identities And Similarities (SIAS) tool (http://imed.med.ucm.es/Tools/sias.html) with BLOSUM 62 as the scoring matrix. The length of the MSA was used as the sequence length denominator used in the percent identity equation.
Cis-regulatory elements data search
Promoter elements present in the sequence of the investigated splicing-related kinases were searched using AtCisDB. Arabidopsis gene ID (AGI) of each splicing-related kinase was input into the AtCisDB database ([68] https://agris-knowledgebase.org/AtcisDB/).
Developmental, abiotic, and photoperiod transcript expression analysis
Relative expression levels of slicing kinases were acquired with Hierarchical Clustering tool from GENEVESTIGATOR database (https://genevestigator.com/; [83]). BAR Arabidopsis eFP browser database for extracting microarray transcript expression data (https://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi; [94]) was mined for developmental and abiotic stress transcriptional changes. Absolute values, which are the raw values provided by BAR, were log2 transformed. Abiotic stress values were normalized against the control. Photoperiod transcript changes were mined using DiurnalDb [http://diurnal.mocklerlab.org/diurnal_data_finders/new; [123]).
Plant growth
Sterilized A. thaliana wild type Col-0 seeds (source ABRC; abrc.osu.edu/) were plated on 0.5 × MS (Caisson Laboratories inc. Murashige & Skoog MSP01-50LT) 0.8 % plant agar (Caisson laboratories inc. Phytoblend™ PTP01-2 KG) and stratified for 3 days at 4 °C in the dark (Supplemental Figure 7). Seeds were germinated and grow for 7 days under 8 h: 16 h, 12 h: 12 h or 16 h: 8 h photoperiod. Seedlings were grown vertically in custom 3D printed blacked-out vertical plate holders to minimize root exposure to light. For 24 h: 0 h photoperiod, seedlings were transferred from 12 h: 12 h after 5 days of entrainment to 24 h: 0 h for 2 days. Plant tissue was harvested at ZT6, ZT11, ZT18 and ZT23. All seedlings were grown under LED light with 100 µmol/m2/s at constant 22 oC with 50 % humidity.
Abiotic stress experimentation
All abiotic stress seedlings were grown in 12 h: 12 h photoperiod. Cold treated seedlings were placed in 4 °C thermo-regulated vertical plate holder at ZT 9 on day 6, then harvested at ZT9 on day 7. Heat treated seedlings were transferred at ZT1 on day 7 to a 38 °C thermo-regulated vertical place holder for 3 h, then placed back at 22 °C for a 3 h recovery time at which point seedlings were harvested at ZT7. Osmotic and salt stressed seedling were carefully transferred at ZT6 from germination media to 300 mM mannitol and 150 mM NaCl to simulate osmotic and salt stress, respectively. Seedlings were exposed for 24 h and harvested the subsequent day at ZT6. All seedlings were grown under LED light with 100 µmol/m2/s at constant 22 °C with 50 % humidity (Supplemental Figure 7).
Tissue processing for transcript quantification
Four biological replicates each containing ~ 100 mg of plant tissue were flash frozen and ground using Geno/Grinder® for 30 s at 1200 rpm. Total RNA was extracted using a modified TRizol protocol [139]. 1 ml of TRI Reagent® (Sigma-Aldrich T9424) was added to each 100 mg of tissue and incubated for 10 min at room temperature. Extracellular material was removed by centrifuging at 13,000 × g for 10 min. Supernatant was transferred to new tubes and 200 µL of chloroform was added. Tubes were then inverted variously for 15 s and then left at RT for 3 min. Phase separation was achieved by centrifuging at 13,000 × g for 15 min at 4 °C. The aqueous phase was transferred to new tubes carefully avoiding the interphase. RNA was precipitated by adding 500 µL of 100 % isopropanol and then left to incubate for 10 min at RT. RNA was pelleted by centrifuging at 13,000 × g for 15 min at 4 °C. Supernatant was removed and pellet was washed with 1 ml of 75% Ethanol. Tube were centrifuged once again at 7500 × g for 5 min at 4 °C. Pellet was dried and subsequently resuspended in nuclease-free water. Total RNA was quantified by NanoDrop ND 1000 spectrophotometer. A minimum of 150 ng of purified RNA in 7.5 µL was sent to NanoString (NanoString Technologies, Seattle, USA, https://www.nanostring.com) for analysis. The NanoString probes were designed and synthesized by NanoString (Supplemental Table 8). PP2AA3 (AT1G13320) and UBQ10 (AT4G05320) were used as reference genes (Czechowski et al., 2005; Hong et al., 2010). All data was normalized the corresponding abundance of PP2AA3 for each sample, with Log2 fold change (FC) calculated between control and stress conditions, with the corrected p-value (FDR) calculated using Benjamini-Yekutieli. PP2AA3 (AT1G13320) and UBQ10 (AT4G05320) were used as reference genes [140, 141]. ZT6 12 h: 12 h tissue was used as a control comparison during data analysis. Positive control genes were selected for each abiotic stress and photoperiod. COR78 (AT5G52310) for cold, HOP3 (AT4G12400) for heat, LEA4-5 (AT5G06760) for osmotic, salt and cold, and KIN1 (AT5G15960) for osmotic, salt, and cold (Supplemental Figure 8). CCA1 (AT2G46830) and TOC1 (AT5G61380) were used for photoperiod (Supplemental Figure 9).
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Change history
23 May 2023
A Correction to this paper has been published: https://doi.org/10.1186/s12870-023-04289-6
References
Bludau I, Aebersold R. Proteomic and interactomic insights into the molecular basis of cell functional diversity. Nat Rev Mol Cell Biol. 2020;21:327–40.
Bludau I, Frank M, Dörig C, Cai Y, Heusel M, Rosenberger G, et al. Systematic detection of functional proteoform groups from bottom-up proteomic datasets. Nat Commun. 2021;12:3810.
Drechsel G, Kahles A, Kesarwani AK, Stauffer E, Behr J, Drewe P, et al. Nonsense-mediated decay of alternative precursor mRNA splicing variants is a major determinant of the Arabidopsis steady state transcriptome. Plant Cell. 2013;25:3726–42.
Filichkin SA, Priest HD, Givan SA, Shen R, Bryant DW, Fox SE, et al. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010;20:45–58.
Filichkin SA, Mockler TC. Unproductive alternative splicing and nonsense mRNAs: A widespread phenomenon among plant circadian clock genes. Biol Direct. 2012;7:1–15.
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–5.
Marquez Y, Brown JWS, Simpson C, Barta A, Kalyna M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012;22:1184–95.
Zhang R, Calixto CPG, Marquez Y, Venhuizen P, Tzioutziou NA, Guo W, et al. A high quality Arabidopsis transcriptome for accurate transcript-level analysis of alternative splicing. Nucleic Acids Res. 2017;45:5061–73.
Martín G, Márquez Y, Mantica F, Duque P, Irimia M. Alternative splicing landscapes in Arabidopsis thaliana across tissues and stress conditions highlight major functional differences with animals. Genome Biol. 2021;22:1–26.
Staiger D, Brown JWS. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell. 2013;25:3640–56.
Carvalho RF, Feijão CV, Duque P. On the physiological significance of alternative splicing events in higher plants. Protoplasma. 2013;250:639–50.
Leister D, Wang L, Kleine T. Organellar gene expression and acclimation of plants to environmental stress. Front Plant Sci. 2017;8:387.
Szakonyi D, Duque P. Alternative splicing as a regulator of early plant development. Front Plant Sci. 2018;9:1–9.
Kanno T, Venhuizen P, Wen TN, Lin WD, Chiou P, Kalyna M, et al. PRP4KA, a putative spliceosomal protein kinase, is important for alternative splicing and development in arabidopsis Thaliana. Genetics. 2018;210:1267–85.
Tognacca RS, Servi L, Hernando CE, Saura-Sanchez M, Yanovsky MJ, Petrillo E, et al. Alternative Splicing Regulation During Light-Induced Germination of Arabidopsis thaliana Seeds. Front Plant Sci. 2019;10:1–12.
McClung CR, Lou P, Hermand V, Kim JA. The importance of ambient temperature to growth and the induction of flowering. Front Plant Sci. 2016;7:1–7.
Chang P, Hsieh HY, Tu SL. The U1 snRNP component RBP45d regulates temperature-responsive flowering in Arabidopsis. Plant Cell. 2022;34:834–51.
Laloum T, Martín G, Duque P. Alternative Splicing Control of Abiotic Stress Responses. Trends Plant Sci. 2018;23:140–50.
Ling Y, Mahfouz MM, Zhou S. Pre-mRNA alternative splicing as a modulator for heat stress response in plants. Trends Plant Sci. 2021;26:1153–70.
Feng J, Li J, Gao Z, Lu Y, Yu J, Zheng Q, et al. SKIP Confers Osmotic Tolerance during Salt Stress by Controlling Alternative Gene Splicing in Arabidopsis. Mol Plant. 2015;8:1038–52.
Ling Y, Alshareef S, Butt H, Lozano-Juste J, Li L, Galal AA, et al. Pre-mRNA splicing repression triggers abiotic stress signaling in plants. Plant J. 2017;89:291–309.
Zhu FY, Chen MX, Ye NH, Shi L, Ma KL, Yang JF, et al. Proteogenomic analysis reveals alternative splicing and translation as part of the abscisic acid response in Arabidopsis seedlings. Plant J. 2017;91:518–33.
Romanowski A, Yanovsky MJ. Circadian rhythms and post-transcriptional regulation in higher plants. Front Plant Sci. 2015;6:1–11.
Grundy J, Stoker C, Carré IA. Circadian regulation of abiotic stress tolerance in plants. Front Plant Sci. 2015;6:1–15.
Wahl MC, Will CL, Lührmann R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell. 2009;136:701–18.
Reddy ASN, Ali GS, Golovkin M. Arabidopsis U1 snRNP 70K protein and its interacting proteins: nuclear localization and in vivo dynamics of a novel plant-specific serine/arginine-rich protein. Symp Soc Exp Biol. 2004;:279–95.
Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, Qiu J, et al. Genome-wide Analysis Reveals SR Protein Cooperation and Competition in Regulated Splicing. Mol Cell. 2013;50:223–35.
Risso G, Pelisch F, Quaglino A, Pozzi B, Srebrow A. Regulating the regulators: Serine/arginine-rich proteins under scrutiny. IUBMB Life. 2012;64:809–16.
Syed NH, Kalyna M, Marquez Y, Barta A, Brown JWS. Alternative splicing in plants - coming of age. Trends Plant Sci. 2012;17:616–23.
Barta A, Kalyna M, Reddy ASN. Implementing a rational and consistent nomenclature for serine/arginine-rich protein splicing factors (SR proteins) in plants. Plant Cell. 2010;22:2926–9.
Zhou Z, Fu XD. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma. 2013;122:191–207.
De La Fuente Van Bentem S, Anrather D, Roitinger E, Djamei A, Hufnagl T, Barta A, et al. Phosphoproteomics reveals extensive in vivo phosphorylation of Arabidopsis proteins involved in RNA metabolism. Nucleic Acids Res. 2006;34:3267–78.
Willems P, Horne A, Van Parys T, Goormachtig S, De Smet I, Botzki A, et al. The Plant PTM Viewer, a central resource for exploring plant protein modifications. Plant J. 2019;99:752–62.
Xue H, Zhang Q, Wang P, Cao B, Jia C, Cheng B, et al. qPTMplants: an integrative database of quantitative post-translational modifications in plants. Nucleic Acids Res. 2022;50:D1491–9.
Tenenbaum SA, Aguirre-Ghiso J. Dephosphorylation shows SR proteins the way out. Mol Cell. 2005;20:499–501.
Gui JF, Lane WS, Fu XD. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994;369:678–82.
Aubol BE, Plocinik RM, Hagopian JC, Ma CT, McGlone ML, Bandyopadhyay R, et al. Partitioning RS domain phosphorylation in an SR protein through the CLK and SRPK protein kinases. J Mol Biol. 2013;425:2894–909.
Velazquez-Dones A, Hagopian JC, Ma CT, Zhong XY, Zhou H, Ghosh G, et al. Mass spectrometric and kinetic analysis of ASF/SF2 phosphorylation by SRPK1 and Clk/Sty. J Biol Chem. 2005;280:41761–8.
Keshwani MM, Aubol BE, Fattet L, Ma CT, Qiu J, Jennings PA, et al. Conserved proline-directed phosphorylation regulates SR protein conformation and splicing function. Biochem J. 2015;466:311–22.
Schneider M, Hsiao HH, Will CL, Giet R, Urlaub H, Lührmann R. Human PRP4 kinase is required for stable tri-snRNP association during spliceosomal B complex formation. Nat Struct Mol Biol. 2010;17:216–21.
Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, et al. The Akt-SRPK-SR Axis Constitutes a Major Pathway in Transducing EGF Signaling to Regulate Alternative Splicing in the Nucleus. Mol Cell. 2012;47:422–33.
Aubol BE, Wozniak JM, Fattet L, Gonzalez DJ, Adams JA. CLK1 reorganizes the splicing factor U1–70K for early spliceosomal protein assembly. Proc Natl Acad Sci U S A. 2021;118:e2018251118.
Giannakouros T, Nikolakaki E, Mylonis I, Georgatsou E. Serine-arginine protein kinases: A small protein kinase family with a large cellular presence. FEBS J. 2011;278:570–86.
Nieratschker V, Schubert A, Jauch M, Bock N, Bucher D, Dippacher S, et al. Bruchpilot in ribbon-like axonal agglomerates, behavioral defects, and early death in SRPK79D kinase mutants of Drosophila. PLoS Genet. 2009;5:e1000700.
Loh BJ, Cullen CF, Vogt N, Ohkura H. The conserved kinase SRPK regulates karyosome formation and spindle microtubule assembly in Drosophila oocytes. J Cell Sci. 2012;125:4457–62.
Clark JW, Donoghue PCJ. Whole-Genome Duplication and Plant Macroevolution. Trends Plant Sci. 2018;23:933–45.
Edger PP, Heidel-Fischer HM, Bekaert M, Rota J, Glöckner G, Platts AE, et al. The butterfly plant arms-race escalated by gene and genome duplications. Proc Natl Acad Sci U S A. 2015;112:8362–6.
De Smet R, Adams KL, Vandepoele K, Van Montagu MCE, Maere S, Van De Peer Y. Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc Natl Acad Sci U S A. 2013;110:2898–903.
Li Z, Defoort J, Tasdighian S, Maere S, Van De Peer Y, De Smet R. Gene duplicability of core genes is highly consistent across all angiosperms. Plant Cell. 2015;28:326–44.
Nakagawa O, Arnold M, Nakagawa M, Hamada H, Shelton JM, Kusano H, et al. Centronuclear myopathy in mice lacking a novel muscle-specific protein kinase transcriptionally regulated by MEF2. Genes Dev. 2005;19:2066–77.
Kuroyanagi N, Onogi H, Wakabayashi T, Hagiwara M. Novel SR-protein-specific kinase, SRPK2, disassembles nuclear speckles. Biochem Biophys Res Commun. 1998;242:357–64.
Grosso AR, Gomes AQ, Barbosa-Morais NL, Caldeira S, Thorne NP, Grech G, et al. Tissue-specific splicing factor gene expression signatures. Nucleic Acids Res. 2008;36:4823–32.
Kannan N, Neuwald AF. Evolutionary constraints associated with functional specificity of the CMGC protein kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2α. Protein Sci. 2004;13:2059–77.
Siebel CW, Feng L, Guthrie C, Fu XD. Conservation in budding yeast of a kinase specific for SR splicing factors. Proc Natl Acad Sci U S A. 1999;96:5440–5.
Forslund K, Pekkari I, Sonnhammer ELL. Domain architecture conservation in orthologs. BMC Bioinformatics. 2011;12:326.
Gentry MS, Pace RM. Conservation of the glucan phosphatase laforin is linked to rates of molecular evolution and the glucan metabolism of the organism. BMC Evol Biol. 2009;9:1–14.
Schläpfer P, Mehta D, Ridderikhoff C, Uhrig RG. DomainViz: Intuitive visualization of consensus domain distributions across groups of proteins. Nucleic Acids Res. 2021;49:W169–73.
Ding J-H, Zhong X-Y, Hagopian JC, Cruz MM, Ghosh G, Feramisco J, et al. Regulated Cellular Partitioning of SR Protein-specific Kinases in Mammalian Cells. Mol Biol Cell. 2006;17:876–85.
Ngo JCK, Giang K, Chakrabarti S, Ma CT, Huynh N, Hagopian JC, et al. A Sliding Docking Interaction Is Essential for Sequential and Processive Phosphorylation of an SR Protein by SRPK1. Mol Cell. 2008;29:563–76.
Sigala I, Koutroumani M, Koukiali A, Giannakouros T, Nikolakaki E. Nuclear Translocation of SRPKs Is Associated with 5-FU and Cisplatin Sensitivity in HeLa and T24 Cells. Cells. 2021;10:1–22.
Koutroumani M, Papadopoulos GE, Vlassi M, Nikolakaki E, Giannakouros T. Evidence for disulfide bonds in SR Protein Kinase 1 (SRPK1) that are required for activity and nuclear localization. PLoS ONE. 2017;12:1–21.
Ghosh G, Adams JA. Phosphorylation Mechanism and Structure of Serine-Arginine Protein Kinases. Febs J. 2011;278:587–97.
LaRonde-LeBlanc N, Guszczynski T, Copeland T, Wlodawer A. Structure and activity of the atypical serine kinase Rio1. Febs J. 2005;272:3698–713.
Vanrobays E, Gelugne J-P, Gleizes P-E, Caizergues-Ferrer M. Late Cytoplasmic Maturation of the Small Ribosomal Subunit Requires RIO Proteins in Saccharomyces cerevisiae. Mol Cell Biol. 2003;23:2083–95.
Kakuta Y, Pedersen LG, Carter CW, Negishi M, Pedersen LC. Crystal structure of estrogen sulphotransferase. Nat Struct Biol. 1997;4:904–8.
Bousquet I, Dujardin G, Slonimski PP. ABC1, a novel yeast nuclear gene has a dual function in mitochondria: It suppresses a cytochrome b mRNA translation defect and is essential for the electron transfer in the bc1 complex. EMBO J. 1991;10:203–31.
Macinga DR, Cook GM, Poole RK, Rather PN. Identification and characterization of aarF, a locus required for production of ubiquinone in Providencia stuartii and Escherichia coli and for expression of 2’-N-acetyltransferase in P. stuartii. J Bacteriol. 1998;180:128–35.
Davuluri RV, Sun H, Palaniswamy SK, Matthews N, Molina C, Kurtz M, et al. AGRIS: Arabidopsis Gene Regulatory Information Server, an information resource of Arabidopsis cis-regulatory elements and transcription factors. BMC Bioinformatics. 2003;4:1–11.
Raventós D, Jensen AB, Rask M-B, Casacuberta JM, John M, San SB. A 20 bp cis-acting element is both necessary and sufficient to mediate elicitor response of maize PRms gene. Plant J. 1995;7:147–55.
Rushton PJ, Torres JT, Parniske M, Wernert P, Hahlbrock K, Somssich IE. Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes. EMBO J. 1996;15:5690–700.
Wang Z, Yang P, Fan B, Chen Z. An oligo selection procedure for identification of sequence-specific DNA-binding activities associated with the plant defence response. Plant J. 1998;16:515–22.
Dhatterwal P, Basu S, Mehrotra S, Mehrotra R. Genome wide analysis of W-box element in Arabidopsis thaliana reveals TGAC motif with genes down regulated by heat and salinity. Sci Rep. 2019;9:1–8.
Punzo P, Grillo S, Batelli G. Alternative splicing in plant abiotic stress responses. 2020;48:2117–26.
Mastrangelo AM, Marone D, Laidò G, De Leonardis AM, De Vita P. Alternative splicing: Enhancing ability to cope with stress via transcriptome plasticity. Plant Sci. 2012;185–186:40–9.
Sureshkumar S, Dent C, Seleznev A, Tasset C, Balasubramanian S. Nonsense-mediated mRNA decay modulates FLM-dependent thermosensory flowering response in Arabidopsis. Nat Plants. 2016;2:1–7.
Gil KE, Park MJ, Lee HJ, Park YJ, Han SH, Kwon YJ, et al. Alternative splicing provides a proactive mechanism for the diurnal CONSTANS dynamics in Arabidopsis photoperiodic flowering. Plant J. 2017;89:128–40.
Ahrazem O, Rubio-Moraga A, Argandoña-Picazo J, Castillo R, Gómez-Gómez L. Intron retention and rhythmic diel pattern regulation of carotenoid cleavage dioxygenase 2 during crocetin biosynthesis in saffron. Plant Mol Biol. 2016;91:355–74.
Leviatan N, Alkan N, Leshkowitz D, Fluhr R. Genome-Wide Survey of Cold Stress Regulated Alternative Splicing in Arabidopsis thaliana with Tiling Microarray. PLoS One. 2013;8:e66511.
Liu J, Sun N, Liu M, Liu J, Du B, Wang X, et al. An autoregulatory loop controlling Arabidopsis HsfA2 expression: Role of heat shock-induced alternative splicing. Plant Physiol. 2013;162:512–21.
Ling Y, Serrano N, Gao G, Atia M, Mokhtar M, Woo YH, et al. Thermopriming triggers splicing memory in Arabidopsis. J Exp Bot. 2018;69:2659–75.
Xia W, Liu R, Zhang J, Mason AS, Li Z, Gong S, et al. Alternative splicing of flowering time gene FT is associated with halving of time to flowering in coconut. Sci Rep. 2020;10:1–11.
Dantas LLB, Calixto CPG, Dourado MM, Carneiro MS, Brown JWS, Hotta CT. Alternative Splicing of Circadian Clock Genes Correlates With Temperature in Field-Grown Sugarcane. Front Plant Sci. 2019;10:1–15.
Zimmermann P, Hirsch-Hoffmann M, Hennig L, Gruissem W. GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol. 2004;136:2621–32.
Kilian J, Whitehead D, Horak J, Wanke D, Weinl S, Batistic O, et al. The AtGenExpress global stress expression data set: Protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 2007;50:347–63.
Kwon Y-J, Park M-J, Kim S-G, Baldwin IT, Park C-M. Alternative splicing and nonsense-mediated decay of circadian clock genes under environmental stress conditions in Arabidopsis. BMC Plant Biol. 2014;14:136.
Wang HY, Lin W, Dyck JA, Yeakley JM, Songyang Z, Cantley LC, et al. SRPK2: A differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J Cell Biol. 1998;140:737–50.
Tremousaygue D, Manevski A, Bardet C, Lescure N, Lescure B. Plant interstitial telomere motifs participate in the control of gene expression in root meristems. Plant J. 1999;20:553–61.
Manevski A, Bertoni G, Bardet C, Tremousaygue D, Lescure B. In synergy with various cis-acting elements, plant insterstitial telomere motifs regulate gene expression in Arabidopsis root meristems. FEBS Lett. 2000;483:43–6.
Gaspin C, Rami JF, Lescure B. Distribution of short interstitial telomere motifs in two plant genomes: Putative origin and function. BMC Plant Biol. 2010;10:283.
Ransom-Hodgkins WD. The application of expression analysis in elucidating the eukaryotic elongation factor one alpha gene family in Arabidopsis thaliana. Mol Genet Genomics. 2009;281:391–405.
Abe M, Takahashi T, Komeda Y. Identification of a cis-regulatory element for L1 layer-specific gene expression, which is targeted by an L1-specific homeodomain protein. Plant J. 2001;26:487–94.
Bustos F, Segarra-Fas A, Nardocci G, Cassidy A, Antico O, Davidson L, et al. Functional Diversification of SRSF Protein Kinase to Control Ubiquitin-Dependent Neurodevelopmental Signaling. Dev Cell. 2020;55:629-647.e7.
Gou LT, Lim DH, Ma W, Aubol BE, Hao Y, Wang X, et al. Initiation of Parental Genome Reprogramming in Fertilized Oocyte by Splicing Kinase SRPK1-Catalyzed Protamine Phosphorylation. Cell. 2020;180:1212–27.
Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, et al. A gene expression map of Arabidopsis thaliana development. Nat Genet. 2005;37:501–6.
Hu YX, Wang YH, Liu XF, Li JY. Arabidopsis RAV1 is down-regulated by brassinosteroid and may act as a negative regulator during plant development. Cell Res. 2004;14:8–15.
Deshmukh V, O’Green AL, Bossard C, Seo T, Lamangan L, Ibanez M, et al. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthr Cartil. 2019;27:1347–60.
Clevers H. Wnt/β-Catenin Signaling in Development and Disease. Cell. 2006;127:469–80.
Cesana M, Guo MH, Cacchiarelli D, Wahlster L, Barragan J, Doulatov S, et al. A CLK3-HMGA2 Alternative Splicing Axis Impacts Human Hematopoietic Stem Cell Molecular Identity throughout Development. Cell Stem Cell. 2018;22:575-588.e7.
Bao X, Franks RG, Levin JZ, Liu Z. Repression of AGAMOUS by BELLRINGER in floral and inflorescence meristems. Plant Cell. 2004;16:1478–89.
Lamb RS, Hill TA, Tan QKG, Irish VF. Regulation of APETALA3 floral homeotic gene expression by meristem identity genes. Development. 2002;129:2079–86.
Li SB, Xie ZZ, Hu CG, Zhang JZ. A review of auxin response factors (ARFs) in plants. Front Plant Sci. 2016;7:1–7.
Rademacher EH, Lokerse AS, Schlereth A, Llavata-Peris CI, Bayer M, Kientz M, et al. Different Auxin Response Machineries Control Distinct Cell Fates in the Early Plant Embryo. Dev Cell. 2012;22:211–22.
Guilfoyle TJ, Hagen G. Auxin response factors. Curr Opin Plant Biol. 2007;10:453–60.
Ellis CM, Nagpal P, Young JC, Hagen G, Guilfoyle TJ, Reed JW. AUXIN RESPONSE FACTOR1 and AUXIN RESPONSE FACTOR2 regulate senescence and floral organ abscission in Arabidopsis thaliana. Development. 2005;132:4563–74.
Srinivasan A, Jiménez-Gómez JM, Fornara F, Soppe WJJ, Brambilla V. Alternative splicing enhances transcriptome complexity in desiccating seeds. J Integr Plant Biol. 2016;58:947–58.
Penfield S, Josse EM, Halliday KJ. A role for an alternative splice variant of PIF6 in the control of Arabidopsis primary seed dormancy. Plant Mol Biol. 2010;73:89–95.
Wang HL, Zhang Y, Wang T, Yang Q, Yang Y, Li Z, et al. An alternative splicing variant of PtRD26 delays leaf senescence by regulating multiple NAC transcription factors in Populus. Plant Cell. 2021;33:1594–614.
Kreps JA, Wu Y, Chang HS, Zhu T, Wang X, Harper JF. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002;130:2129–41.
Kim SY, Chung H-J, Thomas TL. Isolation of a novel class of bZIP transcription factors that interact with ABA-responsive and embryo-specification elements in the Dc3 promoter using a modified yeast one-hybrid system. Plant J. 1997;11:1237–51.
Chon W, Provart NJ, Glazebrook J, Katagiri F, Chang HS, Eulgem T, et al. Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell. 2002;14:559–74.
Kim JS, Mizoi J, Yoshida T, Fujita Y, Nakajima J, Ohori T, et al. An ABRE promoter sequence is involved in osmotic stress-responsive expression of the DREB2A gene, which encodes a transcription factor regulating drought-inducible genes in Arabidopsis. Plant Cell Physiol. 2011;52:2136–46.
Vivekananda J, Drew MC, Thomas TL. Hormonal and environmental regulation of the carrot lea-class gene Dc3. Plant Physiol. 1992;100:576–81.
Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev. 2009;23:482–95.
Vivarelli S, Lenzken SC, Ruepp MD, Ranzini F, Maffioletti A, Alvarez R, et al. Paraquat Modulates Alternative Pre-mRNA Splicing by Modifying the Intracellular Distribution of SRPK2. PLoS One. 2013;8:e61980.
Haltenhof T, Kotte A, De Bortoli F, Schiefer S, Meinke S, Emmerichs AK, et al. A Conserved Kinase-Based Body-Temperature Sensor Globally Controls Alternative Splicing and Gene Expression. Mol Cell. 2020;78:57-69.e4.
Groß T, Lützelberger M, Wiegmann H, Klingenhoff A, Shenoy S, Käufer NF. Functional analysis of the fission yeast Prp4 protein kinase involved in pre-mRNA splicing and isolation of a putative mammalian homologue. Nucleic Acids Res. 1997;25:1028–35.
Rosenberg GH, Alahari SK, Käufer NF. prp4 from Schizosaccharomyces pombe, a mutant deficient in pre-mRNA splicing isolated using genes containing artificial introns. MGG Mol Gen Genet. 1991;226:305–9.
Perez-santángelo S, Schlaen RG, Yanovsky MJ. Genomic analysis reveals novel connections between alternative splicing and circadian regulatory networks. Brief Funct Genomics. 2013;12:13–24.
Rook F, Gerrits N, Kortstee A, Van Kampen M, Borrias M, Weisbeek P, et al. Sucrose-specific signalling represses translation of the Arabidopsis ATB2 bZIP transcription factor gene. Plant J. 1998;15:253–63.
Satoh R, Fujita Y, Nakashima K, Shinozaki K, Yamaguchi-Shinozaki K. A novel subgroup of bZIP proteins functions as transcriptional activators in hypoosmolarity-responsive expression of the ProDH gene in Arabidopsis. Plant Cell Physiol. 2004;45:309–17.
Hudson ME, Quail PH. Identification of Promoter Motifs Involved in the Network of Phytochrome A-Regulated Gene Expression by Combined Analysis of Genomic Sequence and Microarray Data. Plant Physiol. 2003;133:1605–16.
Park S, Lee Y, Lee JH, Jin ES. Expression of the high light-inducible Dunaliella LIP promoter in Chlamydomonas reinhardtii. Planta. 2013;238:1147–56.
Mockler TC, Michael TP, Priest HD, Shen R, Sullivan CM, Givan SA, et al. The diurnal project: Diurnal and circadian expression profiling, model-based pattern matching, and promoter analysis. Cold Spring Harb Symp Quant Biol. 2007;72:353–63.
Song Y, Jiang Y, Kuai B, Li L. Circadian clock-associated 1 inhibits leaf senescence in arabidopsis. Front Plant Sci. 2018;9:1–9.
James AB, Syed NH, Bordage S, Marshall J, Nimmo GA, Jenkins GI, et al. Alternative splicing mediates responses of the Arabidopsis circadian clock to temperature changes. Plant Cell. 2012;24:961–81.
Seo PJ, Park MJ, Lim MH, Kim SG, Lee M, Baldwin IT, et al. A self-regulatory circuit of CIRCADIAN CLOCK-ASSOCIATED1 underlies the circadian clock regulation of temperature responses in Arabidopsis. Plant Cell. 2012;24:2427–42.
Ali GS, Golovkin M, Reddy ASN. Nuclear localization and in vivo dynamics of a plant-specific serine/arginine-rich protein. Plant J. 2003;36:883–93.
Golovkin M, Reddy ASN. An SC35-like protein and a novel serine/arginine-rich protein interact with Arabidopsis U1–70K protein. J Biol Chem. 1999;274:36428–38.
James AB, Syed NH, Brown JWS, Nimmo HG. Thermoplasticity in the plant circadian clock. Plant Signal Behav. 2012;7:1219–23.
Papoutsopoulou S, Nikolakaki E, Chalepakis G, Kruft V, Chevaillier P, Giannakouros T. SR protein-specific kinase 1 is highly expressed in testis and phosphorylates protamine 1. Nucleic Acids Res. 1999;27:2972–80.
Sridhara SC, Carvalho S, Grosso AR, Gallego-Paez LM, Carmo-Fonseca M, de Almeida SF. Transcription Dynamics Prevent RNA-Mediated Genomic Instability through SRPK2-Dependent DDX23 Phosphorylation. Cell Rep. 2017;18:334–43.
Takano M, Koyama Y, Ito H, Hoshino S, Onogi H, Hagiwara M, et al. Regulation of Binding of Lamin B Receptor to Chromatin by SR Protein Kinase and cdc2 Kinase in Xenopus Egg Extracts. J Biol Chem. 2004;279:13265–71.
Jang SW, Yang SJ, Ehlén Å, Dong S, Khoury H, Chen J, et al. Serine/arginine protein-specific kinase 2 promotes leukemia cell proliferation by phosphorylating acinus and regulating cyclin A1. Cancer Res. 2008;68:4559–70.
Katoh K, Misawa K, Kuma KI, Miyata T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66.
Leebens-Mack JH, Barker MS, Carpenter EJ, Deyholos MK, Gitzendanner MA, Graham SW, et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature. 2019;574:679–85.
Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016;44:W232–5.
Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9.
Hong SM, Bahn SC, Lyu A, Jung HS, Ahn JH. Identification and testing of superior reference genes for a starting pool of transcript normalization in Arabidopsis. Plant Cell Physiol. 2010;51:1694–706.
Czechowski T, Stitt M, Altmann T, Michael K, Udvardi A, Scheible W-R. Genome-wide Identification and Testing of Superior Reference Genes for Transcript normalization in Arabidopsis. Plant Physiol. 2005;139:5–17.
Acknowledgements
The authors would like to thank Dr. Rhiannon Peery for helpful discussions on our phylogenetic analysis, and Dr. Anthony Cornish of the Molecular Biology Services Unit (MBSU) for technical help with the abiotic stress treatments.
Funding
This funding was supported by the National Science and Engineering Research Council of Canada (NSERC).
Author information
Authors and Affiliations
Contributions
The study was conceived of by MCRG and RGU. MCRG performed all phylogenetic tree analysis, along with ePlant and AtCisDB analyses. MCRG also performed all plant growth, harvesting, RNA isolation for the transcriptomic aspects of the manuscript in addition to all subsequent data analysis. QL performed the genevestigator analysis and assisted with data analysis. DM assisted with data analysis and visualization as well as manuscript assembly, while MCRG wrote the manuscript with writing, assembly and editorial support from RGU. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: the authors identified an error in the author name of D. Mehta. The author name was not captured correctly.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rodriguez Gallo, M.C., Li, Q., Mehta, D. et al. Genome-scale analysis of Arabidopsis splicing-related protein kinase families reveals roles in abiotic stress adaptation. BMC Plant Biol 22, 496 (2022). https://doi.org/10.1186/s12870-022-03870-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03870-9