
Abstract
In contrast to most land plant species, sorbitol, instead of sucrose, is the major photosynthetic product in many Rosaceae species. It has been well illustrated that three key functional genes encoding sorbitol-6-phosphate dehydrogenase (S6PDH), sorbitol dehydrogenase (SDH), and sorbitol transporter (SOT), are mainly responsible for the synthesis, degradation and transportation of sorbitol. In this study, the genome-wide identification of S6PDH, SDH and SOT genes was conducted in four Rosaceae species, peach, mei, apple and pear, and showed the sorbitol bio-pathway to be dominant (named sorbitol present group, SPG); another three related species, including tomato, poplar and Arabidopsis, showed a non-sorbitol bio-pathway (named sorbitol absent group, SAG). To understand the evolutionary differences of the three important gene families between SAG and SPG, their corresponding gene duplication, evolutionary rate, codon bias and positive selection patterns have been analyzed and compared. The sorbitol pathway genes in SPG were found to be expanded through dispersed and tandem gene duplications. Branch-specific model analyses revealed SDH and S6PDH clade A were under stronger purifying selection in SPG. A higher frequency of optimal codons was found in S6PDH and SDH than that of SOT in SPG, confirming the purifying selection effect on them. In addition, branch-site model analyses revealed SOT genes were under positive selection in SPG. Expression analyses showed diverse expression patterns of sorbitol-related genes. Overall, these findings provide new insights in the evolutionary characteristics for the three key sorbitol metabolism-related gene families in Rosaceae and other non-sorbitol dominant pathway species.
Similar content being viewed by others


Introduction
Sugars, are important compounds in all plants, and play critical roles in both primary and secondary metabolism. Notably, sugars are important in Rosaceae fruit trees, where they accumulate in the fruits, which are the main organs consumed. Such fruit trees, which represent the most important sources of fruit for human consumption, include pear, apple, peach, and mei. Overall, the sugar content in fruit is determined by the carbon partitioning system in plants, a critical process that distributes chemical energy converted by the plant through photosynthesis [1]. Compared with most land plants, the source-sink system in pear and several other Rosaceae species, is different in terms of the type of sugar translocated from source to sink, which was sucrose in the former and sorbitol in the latter [2,3,4]. System evolution analyses of Rosaceae species showed that sorbitol is present in the Spiraeoideae and Dryadoideae subfamilies, whereas it is absent in the Rosoideae subfamily [5]. The soluble sugars in mature pear fruit comprise fructose, glucose, sucrose and sorbitol [6]. Sorbitol is not only a key metabolite in carbohydrate metabolism, but also a regulatory signal in stamen development, pollen tube growth and resistance response [7,8,9].
The biosynthesis of sorbitol occurs in the cytosol of leaf cells and is different from sucrose in being produced from glucose 6-phosphate in two catalytic steps (Fig. 1). First, NADP-dependent sorbitol-6-phosphate dehydrogenase (S6PDH, EC 1.1.1.200) catalyzes glucose 6-phosphate into sorbitol 6-phosphate [10]. Secondly, sorbitol-6-phosphate phosphatase (SorPP, EC 3.1.3.50) catalyzes sorbitol-6-phosphate into sorbitol [11]. The transportation of both sucrose and sorbitol occurs from leaves to fruits through the phloem, but with different transporters, namely sucrose transporter (SUT) and sorbitol transporter (SOT). In fruits, sorbitol is converted into glucose or fructose by the activity of three enzymes, NADH-dependent sorbitol dehydrogenase (NAD-SDH, EC 1.1.1.14) [12], DADPH-dependent sorbitol dehydrogenase (NADP-SDH, EC 1.1.1.21) [13], and sorbitol oxidase (SOX, EC 1.1.3.x) [14]. In total, there are five enzymes and one transporter that are closely related to the biosynthesis, degradation and transportation of sorbitol in plants. Till now, three key genes, S6PDH, SDH and SOT, which are known to be involved in sorbitol biosynthesis, degradation and transportation, have been well demonstrated in plants [2].

The scheme of sorbitol metabolism pathway for S6PDH, SOT and SDH. Note: SorPP, SOX and NADP-SDH are marked in red as their sequences have not been characterized in plants. G-6-P: glucose-6-phosphate, Sor-6-P: sorbitol-6-phosphate
S6PDH, localized mainly in leaf cytosol and chloroplast, has been reported to play multiple roles in plants, including not only cold, dark, and abscisic acid stresses [15], but also osmotic [16] and salt stresses [17]. SDH, a cytosolic protein required for sorbitol metabolism [18], emerged very early during evolution [19], and plays a role in abiotic stress in Arabidopsis [20] and tomato [21]. For example, overexpression of SDH in Arabidopsis confers tolerance to salt and osmotic stress [22]. In addition, the presence of SOT is correlated with the accumulation of sorbitol under conditions of drought stress in apple [23] and regulates sorbitol accumulation in pear fruit [24, 25]. Also, previous research has shown that S6PDH, SDH and SOT are members of larger gene families in apple [26], pear [27] and peach (Prunus persica) [28] genomes than in other plant genomes. This finding may be due gene duplication, which is an important feature of genome evolution [29].
Therefore, the aim of this study is to reveal the differences between contrasting species, in which the sugar pathway is either dominated by sorbitol or lacks sorbitol, by investigating the key genes S6PDH, SDH and SOT. Since the sugar pathway of pear (Pyrus bretschneideri), apple (Malus domestica), peach (Prunus persica) and mei (Prunus mume) in Rosaceae is dominated by sorbitol, in this study, we designated these species as the sorbitol present group (SPG) group. In contrast, Arabidopsis, a model eudicot plant, poplar (Populus trichocarpa), a model woody plant species, and tomato (Solanum lycopersicum), a model fruit plant, are members of the sorbitol absent group (SAG) group. We identified S6PDH, SDH and SOT genes of the SAG and SPG groups through a cluster of orthologous groups of proteins (COG) method, then created phylogenetic trees for them and performed evolutionary rate and codon usage bias analyses. We compared the evolutionary pattern of different genes in the two groups to determine their individual features. Overall, our study was designed to provide new insights into the evolutionary characteristics for the three key sorbitol metabolism-related gene families in the Rosaceae and other non-sorbitol dominant pathway species.
Materials and methods
Genome resources
Evolutionary analysis was conducted on seven species based on whether sorbitol was present as the major translocated sugar. These species were designated as either the sorbitol present group (SPG) or the sorbitol absent group (SAG). The SPG includes pear (Pyrus bretschneideri), apple (Malus domestica), peach (Prunus persica) and mei (Prunus mume) in Rosaceae and the SAG includes Arabidopsis, poplar (Populus trichocarpa), and tomato (Solanum lycopersicum). The phylogenetic tree of these seven species is shown in Fig. 2. Genome resources of the various species were collected from public databases (Table 1). The apple, peach, tomato, poplar, and Arabidopsis genome sequences were retrieved from Phytozome version 9 (http://www.phytozome.net). The pear genome sequences were retrieved from GigaDB (http://gigadb.org/dataset/100083) and the mei genome sequences were retrieved from the Mei Genome Project website (https://github.com/lileiting/prunusmumegenome).

Phylogenetic tree for seven species used in this study. Note: Right table showed the number of genes
Gene identification
Protein sequences from the selected genomes were first used to build a cluster of orthologous groups (COG) dataset [30,31,32]. This procedure was based on the concept that a family of in-paralogs in one lineage can be orthologous to a single gene in another lineage and orthologs could be identified by the identification of an orthologous family. In such a family, a pair of sequences could be identified as two kinds of relationship, i.e. symmetrical and asymmetrical BeTs (the Best Hits). The orthologous family would form a network after linking the symmetrical and asymmetrical BeTs by solid and broken lines; and thus all the members in the network could be identified with one member investigated [30, 31, 33, 34]. Briefly, there were five steps. First, two types of all-against-all protein sequence comparisons were carried out using PSI-BLAST with and without the SEG filter (low complexity masking), and composition-based score adjustment [35] was carried out. Both methods used the parameter of “-show_gis -outfmt 7 -num_descriptions 1000 -num_alignments 1 000 -dbsize 100000000” as suggested [34]. Secondly, after processing the BLAST results with an E-value threshold of 0.1 using the program COGreadblast, and collecting lineage-specific expansion using the program COGlse, clusters were made from symmetrical best hits using the program COG triangle with an E-value threshold of 0.01 and a hit coverage threshold of 0.5 [34]. Thirdly, three representative proteins of S6PDH, SDH and SOT with status of reviewed were retrieved from the Uniprot database (http://www.uniprot.org/) with accession number of P28475, Q9FJ95, Q8RI1 (Table S1). Fourth, each corresponding locus was queried against COG datasets constructed from the second step and the respective COGs were obtained. Fifth, each sequence in the obtained COG was repeated as in the fourth step, until no new sequence was found. The presence of a specific gene in multiple COGs was allowed, in order to ensure all homologs were included. To further confirm the results, the identified genes were submitted to CDD [36] to determine their protein domains. Results from CDD confirmed their membership of superfamilies; specifically, all S6PDH genes belonged to the aldo-keto reductase (AKR) superfamily (cl00470), all SDH genes belonged to the medium-chain dehydrogenases/reductases (MDR) superfamily (cl16912), and all SOT genes belonged to the major facilitator superfamily (MFS, cl21472).
Phylogenetic analysis
The coding sequences of genes in each gene family were aligned using the codon model in PRANK [37]. The aligned sequence was then translated into amino acids and the best substitution models were tested using Prottest 3 [38]. The results showed that for S6PDH, the best evolutionary model implemented in RAxML [39] is LG + G (the substitution matrix [40], and a gamma model of rate heterogeneity), and the best evolutionary model implemented in MrBayes [41] is JTT + G (the substitution matrix [42], and a gamma model of rate heterogeneity). For SDH, the best evolutionary model implemented in RAxML and MrBayes were both JTT + G. For SOT, the two best evolutionary models implemented in RAxML and MrBayes were JTT + I + G (the substitution matrix [42], a proportion of invariant sites, and a gamma model of rate heterogeneity).
Phylogenetic trees were constructed using the Bayesian method implemented in MrBayes [41], running one million generations for each gene family, and discarding the first 25% samples as burn-in. Convergence was assessed by the potential scale reduction factor [43]. Additionally, maximum likelihood trees with 1000 bootstrap replicates using RAxML [39] were reconstructed and bootstrap support values were added to the Baysian trees using SumTrees [44]. Branches in the phylogenetic trees with posterior probabilities less than 0.80 were removed using Dendroscope 3 [45] and phylogenetic trees were visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/). Genes in the SPG were divided into different clades by the distribution of genes in combination with the species tree. Each clade in a gene family typically contains genes from the four SPG species and clades in the same gene family share the common ancestor corresponding to the speciation of SPG species.
Gene duplication type identification
Protein sequences for each of the seven species were independently performed with an all-against-all BLASTP search with E-value threshold of 1e-5 to search for potential anchors between every possible pair of chromosomes. The homologous genes were used as input for the program MCScanX [46] to search collinear blocks and gene types. Four types of gene duplication including dispersed, proximal, tandem and WGD/segmental were assigned by MCScanX.
Pairwise Ka/Ks calculation
The coding sequences of each group of genes was pairwise aligned using PRANK [37], and then the alignment sequences to AXT format were converted and imported into KaKs_Calculator 2.0 [47] to calculate Ka/Ks using YN model. The R programing language (http://www.r-project.org) was used to make the boxplot.
Estimation of branch-specific evolutionary rates and detection of positive selection
The coding sequences of each gene family were aligned using a codon model in PRANK [37]. The evolutionary rate of each gene family and branches for SPG, was estimated using branch-specific model in PAML 4.7a [48]. First, we used a one-ratio model, assuming the evolutionary rate in the whole phylogenetic tree was the same. Secondly, we used a two-ratio model, assuming the evolutionary rate in the SPG genes is different from the SAG genes. Thirdly, we used a multiple-ratio model, assuming each branch representing the speciation of sorbitol present species was independent, and has a different ratio from background ratio. Furthermore, a likelihood ratio test (LRT) was used to test if the two-ratio model rejects the one-ratio model, or the multiple-ratio model rejects the one-ratio models.
To test if those clades of SPG, as marked in Fig. 2 for the three gene families, were subject to positive selection, we used the program Fitmodel [49] to conduct the selection analysis, which was a maximum likelihood-based program used for estimating parameters of sequence evolution. Fitmodel allowed the site-specific selection process to vary along lineages for a phylogenetic tree (switching model). M3 [50] and M3 + 1 [49] models in Fitmodel were employed in this analysis to test positive selection sites.
Codon usage bias analysis
The overall codon bias for all genes in the seven genomes was calculated using the method of effective number of codons (ENC) [51] with the ENCprime package [52]. The measure does not make any assumptions, including optimal codons or GC contents. Values of Nc ranged from 20, for extremely biased genes that use only one codon per amino acid, to 61, for genes that use all synonymous codons equally [51, 53]. Short sequences of less than 50 codons were removed from the analysis. The optimal codon for seven species was determined using a method similar to that of [53]. Briefly, the correlation of codon frequency of each codon in their codon family was calculated with the overall codon bias (Nc). The optimal codon for each codon family was defined as t2he codon that showed the strongest and most significant negative correlation with Nc. Codon families that appeared less than 10 times were removed. The threshold of significance is 0.05/n, where n is the number of codons in the codon family. Spearman correlation was performed using the R programming language.
Frequency of optimal codons (FOP) was defined as the ratio of optimal codons to the sum of non-optimal codons and optimal codons. The formula is as follows: FOP = number of optimal codons / (Number of optimal codons + Number of non-optimal codons). In addition, GC content and GC3 content were calculated using CodonW package (http://codonw.sourceforge.net).
Expression analysis
In total, 20 libraries for apple, mei, pear and peach were retrieved from the SRA database (http://www.ncbi.nlm.nih.gov/sra) and used to perform expression analysis for S6PDH, SDH and SOT genes.
These included five libraries for apple, comprising one leaf library (SRR767660) and four fruit libraries from different developmental stages, 25 daa (days after anthesis), 35 daa, 60 daa and 87 daa (SRR768128, SRR768129, SRR768130, SRR768131); five libraries for mei, comprising bud (SRR542478), leaf (SRR542479), root (SRR542480), stem (SRR542481) and fruit (SRR542482); six libraries for pear fruit (SRR654690, SRR654692, SRR654693, SRR654695, SRR654699, SRR654700); and four libraries for peach, comprising leaf (SRR531862), root (SRR531863), fruit (SRR531864), and embryos and cotyledons (SRR531865).
The downloaded SRA format data were first converted to FASTQ format using the SRA toolkit (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software), then mapped to reference genomes with Tophat v2.1.0 [54] and normalized to fragments per kilobase exons per million reads (FPKM) using Cufflinks v2.2.1 [55] with default parameters. The expression data were subjected to log2 transformation and then visualized using MeV (http://www.tm4.org/mev.html).
Results
Expansion in size of S6PDH, SDH and SOT gene families
The expansion of gene family size (typically by gene duplication) is important for biological evolution by supplying greater genetic diversity (Zhang 2003). To investigate differences in gene family size between the SPG (peach, mei, pear and apple) and the SAG (tomato, Arabidopsis, and poplar), the identified genes in the S6PDH, SDH and SOT families were compared. In total, 20 S6PDH, 33 SDH and 59 SOT genes were identified (Fig. 2, Table S2). In these gene families, the average gene numbers in SPG (4.0 for S6PDH, 7.5 for SDH and 13.75 for SOT) are larger than those in the SAG (1.3 for S6PDH, 1.0 for SDH and 1.3 for SOT), indicating gene family size expansion is contributing to the evolution of the sorbitol character. Although the gene numbers in pear and apple are constantly larger than those in the SAG, this is not the case for the S6PDH and SDH genes in peach and mei, which have similar or identical gene numbers, that is one or two. This indicated that gene family size expansion did not necessarily exist in all three gene families. Only the SOT family was expanded in peach and mei. The expansion in gene family size in pear and apple (Fig. 2) could be partially explained by recent whole genome duplication [26, 27]; for example, the number of SOT genes in the apple genome (15 genes) is about twice that in peach and mei genomes (both have 8 genes), which have no recent whole genome duplications [28, 56].
Distinct evolutionary divergence pattern for S6PDH, SDH and SOT gene families
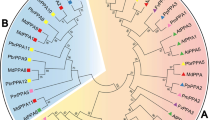
To investigate the phylogenetic relationships of the S6PDH, SDH and SOT gene families in the seven species, we constructed phylogenetic trees (Fig. 3) using Bayesian and maximum likelihood methods, which showed different evolutionary patterns. We found that genes in the SPG were divided into different numbers of clades. Each clade contains genes from four SPG species and different clades of the same gene family share the common ancestor in the ancestral genome of the four SPG species. There were two clades for S6PDH (Fig. 3A), one clade for SDH (Fig. 3B) and three clades for SOT (Fig. 3C) in the SPG. This indicated that the S6PDH and SOT gene families were duplicated, and corresponded with the generation of sorbitol characteristics in Rosaceae. In addition, the number of genes of different clades was uneven with more genes in the S6PDH clade B (10) than clade A (6) (Fig. 3A), and more genes in the SOT clade C (41) than in clades A (6) and B (8) (Fig. 3C). Such clades with more genes may be a consequence of the greater number of gene duplication events. To further illustrate how those genes increased in number, intra-genome synteny analysis (Table S3) was performed. The results showed evidence of distinct gene family expansion. For S6PDH, 13 out of 20 genes were dispersed and duplicated; for SDH, 10 and 11 genes out of 33 SDH genes were dispersed and tandem duplicated; for SOT, 29 and 17 out of 59 SOT genes were dispersed and tandem duplicated, respectively.

Phylogenetic tree of S6PDH, SDH and SOT gene families. A S6PDH gene family, B SDH gene family, C SOT gene family. Note: Posterior probability is labeled in the upper side of each branch and bootstrap support value is labeled in the side of each branch. The gene duplication type is labeled for each gene
Purifying and positive selection in S6PDH, SDH and SOT gene families
To test the selection pressure for S6PDH, SDH and SOT gene families, we performed both branch-specific models of PAML and pairwise Ka/Ks analyses (Fig. 4). For the branch-specific model analyses, we used one-ratio, two-ratio and multiple ratio models (Table 2). The one-ratio model assumed genes on the phylogenetic tree had the same evolutionary ratio, and the results showed that SOT (ω0 = 0.192) had the greatest evolutionary ratio, SDH had the smallest evolutionary ratio (ω0 = 0.104), and evolutionary ratio for S6PDH (ω0 = 0.164) was intermediate. The two-ratio model assumed genes in SPG had the same evolutionary ratio (ω1, foreground evolutionary ratio) but had a different background ratio (ω0). The results showed a higher foreground evolutionary ratio for three gene families, among which ω1 = 0.515 for S6PDH, ω1 = 0.211 for SDH, and ω1 = 0.412 for SOT. All the genes in our study have been through a purifying selection and SDH underwent the strongest purifying selection. In contrast, purifying selection was relaxed in the S6PDH and SOT genes in the SPG. To confirm the results, we performed pairwise Ka/Ks analyses for genes of each gene family in SAG and SPG (Fig. 4). The results showed that the average Ka/Ks value of SDH (average Ka/Ks = 0.113) was smaller than that of S6PDH (average Ka/Ks = 0.241) and SOT (average Ka/Ks = 0.181) in the SPG group. Similarly, stronger purifying selection was observed in the SPG genes. As S6PDH and SOT genes in the SPG have a complex evolutionary history with more than one clade, the multiple-ratio model for branch-specific model analyses was applied to test selection pressure in each clade in S6PDH and SOT. Interestingly, inside the SPG clades, two clades of S6PDH have a very different evolutionary ratio. Clade B had a value (ω2 = 0.629) greater than the background ratio (ω0 = 0.152), while clade A (ω1 = 0.043) was less than the background ratio, which indicated an even stronger purifying selection of S6PDH clade A. The SOT clade C (ω3 = 0.581) had a higher evolutionary ratio than SOT clade A (ω1 = 0.346) and SOT clade B (ω2 = 0.374) (Table 2), indicating the SOT clade C has undergone relaxed purifying selection like S6PDH clade B. Furthermore, a branch-site model was used to investigate whether the three gene families have positive selection after the divergence of sorbitol present species. We first ran M3 and M3 + 1 models in Fitmodel for each branch, and examined them using a likelihood ratio test. The results showed that all the three branches in SOT showed significance by likelihood ratio test (LRT), indicating SOT genes were positively selected in the identified sites. In total, 44 sites in SOT branch A, 8 sites in SOT branch B, and 8 sites in SOT branch C were positively selected.

Comparison of pairwise Ka/Ks values between SAG and SPG for S6PDH, SDH and SOT gene families. Note: sorbitol present group (SPG) and sorbitol absent group (SAG)
Codon bias pattern of SDH is different from S6PDH and SOT
Levels of codon bias are often used as an indicator of the efficiency of purifying selection [57, 58]. To determine the strength of such strong purifying selection in SDH identified above, frequency of optimal codons (FOP) was calculated to explore codon bias in the SAG and SPG genes of three gene families (Fig. 5A and Table S4). Increased FOP indicates increased codon bias. The results showed that, in SAG, SDH genes have the highest average value of FOP, which was consistent with the strongest purifying selection as mentioned in the previous section. The FOP of SOT in both SAG and SGP genes was lowest, which was consistent with the relaxation of purifying selection observed in SOT. As FOP may be related to GC or GC3 bias [58], we also measured the GC and GC3 contents (Fig. 5B and C). The results showed that GC and GC3 contents increased from significantly lower than 50% to around 50% for SDH and SOT in SPG than SAG. This indicated that the relaxation of purifying selection of SDH and SOT in SPG was largely driven by the flexibility of GC and GC3 contents. Since ENC was not affected by the GC and GC3 contents in the same manner as FOP [58], we also measured the ENC values (Fig. 5D). The results also showed the strongest codon bias of SDH in the SAG (by lowest ENC), confirming the strong purifying selection in SDH, irrespective of GC and GC3 bias.

Codon bias pattern analysis between SAG and SPG for S6PDH, SDH and SOT gene families. A Frequency of optimal codons (FOP); B effective number of codon (ENC); C Rate of GC content; D Rate of GC3 content
Tissue-specific expression profiles of S6PDH, SDH and SOT genes in SPG species
To examine the expression pattern of S6PDH, SDH and SOT genes in the four SPG species, we collected and analyzed RNA-seq data from public databases (Fig. 6). The results showed that only a proportion of the genes were expressed in the examined samples and genes were expressed differently in different tissues. Apple genes were examined in one leaf and four fruit tissues (25 days after anthesis (daa), 35 daa, 60 daa, and 87 daa) of ‘Golden Delicious’. The results showed that there were 2 S6PDH, 10 SDH, and 8 SOT genes expressed (FPKM > 0.5) in these tissues (Fig. 6A). Among these genes, MDP0000408705 in S6PDH clade B was specifically highly expressed (FPKM > 1000) in leaf. In addition, some other genes like MDP0000312001 (S6PDH-A), MDP0000250546 (SDH-A) and MDP0000787701 (SOT-B) were also highly expressed in the leaf tissue, indicating their important roles in sorbitol metabolism in this tissue, particularly the accumulation of sugar compounds. Expression levels of four SDH genes (MDP0000188052, MDP0000250546, MDP0000188054, MDP0000874667) were found to be correlated with apple fruit maturation, with increasing expression at four time points of fruit development. Interestingly, the former three of the SDH genes were located in the same sub-clade in the phylogenetic tree (Fig. 3B), indicating coding sequence pattern may play a role in the expression pattern. Additionally, the SOT genes showed temporal expression divergence; some were highly expressed in the first stage of fruit development (like MDP0000296050) and some were highly expressed in the later stage (like MDP0000896307). Pear genes showed similar patterns, in that some of the genes (3 S6PDH, 12 SDH, and 11 SOT) were expressed (FPKM > 0.5) at six stages (S1: 15 days after flowering (daf); S2: 36 daf; S3: 80 daf; S4: 110 daf; S5: 145 daf; S6: 167 daf) of fruit development (Fig. 6B). Two of the S6PDH genes (Pbr042781.1, Pbr023248.1) tended to be expressed in the later stage of fruit development. Some of the SDH genes were highly expressed in almost all fruit developmental stages (Pbr032775.1, Pbr013913.1), some were highly expressed in the early stages only (Pbr013912.1), and some were highly expressed in the later stages (Pbr032776.1), indicating temporal expression divergence in this gene family. In the mei genome, 2 S6PDH, 1 SDH, and 5 SOT genes were found to be expressed in five tissues (bud, leaf, root, stem, and fruit) (Fig. 6C). Among these, Pm017042 (S6PDH-A), Pm019393 (SDH-A), Pm021648 (SOT-A), and Pm021647 (SOT-B) were more highly expressed in the leaf tissue, indicating their important roles in sorbitol accumulation. Additionally, only three genes from the peach genome were found to be expressed in the four tissues (leaf, root, fruit, and embryos and cotyledons), one for each gene family (Fig. 6D).

Expression profiles of S6PDH, SDH and SOT genes in different tissues of apple, pear, mei and peach. A The leaf tissue and four developmental stages of fruits of apple (25 daa (days after anthesis), 35 daa, 60 daa, and 80 daa). B The pear fruit in six developmental stages (S1: 15 days after flowering; S2: 36 days after flowering; S3: 80 days after flowering; S4: 110 days after flowering; S5: 145 days after flowering; S6: 167 days after flowering). C The bud, leaf, root, stem, and fruit tissues of mei. D The leaf, root, fruit, and embryo and cotyledon tissues of peach. Note: The expression data were normalized using fragments per kilobase of exon per million mapped reads (FPKM), with log2 transformed
Discussion
To understand how genes related to sorbitol have evolved in the context of the major photosynthetic product changing from sucrose in most land plants to sorbitol in many species in the Rosaceae, this study first performed comparative analyses of three key genes related to sorbitol metabolism (S6PDH for biosynthesis, SDH for sorbitol degradation, and SOT for sorbitol transportation) in two groups of species, with and without sorbitol as the major photosynthetic product. Although there were some previously published studies related to SDH [59] and SOT [60] genes, they were only focused on either one gene family or one species. Compared with these two previous studies, we found that the number of genes identified in the present study is similar, with minor differences due to identification methodology. The seven species used in the present study were also involved in a previous study that identified SDH genes from 42 angiosperm species [59]; the previous results were similar to those from the present study. However, gene number for five of these species were different, i.e. 2, 3, 4, 16, and 5 genes for poplar, mei, peach, apple and pear in the previous study, but they were 1, 1, 1, 15 and 13 in this study. This was mainly due to the fact that [59] designated L-idonate-5-dehydrogenase (LIDH, EC 1.1.1.264) as SDH Class II and included it in the analyses, while in this study, the LIDH genes were not included since they are not present in the SAG group. Additionally, SDH genes for pear were identified from the literature in [59], rather than from the pear genome resource as in this study. The SOT genes were designated as polyol/monosaccharide transporter (PLT) in the previous study for sugar transporter genes in pear [60], which identified 23 PLT/SOT genes. In this study, the number to 24 SOT genes with the addition of Pbr018904.1. To examine the putative function of Pbr018904.1, we performed a homology search against public databases using NCBI BLAST [61] and confirmed this gene as PLT/SOT.
The sorbitol characteristic was first identified in the Spiraeoideae and Dryadoideae subfamilies but not in the Rosoideae subfamily [5], indicating a stable genetic mechanism was present and had spread in the former subfamilies. Since S6PDH, SDH and SOT were key genes involved in sorbitol metabolism, it is likely that they have undergone evolutionary changes to generate this new character. In this study, we found S6PDH, SDH and SOT have evolved into two, one and three clades (Fig. 3), respectively in SPG. As the formation of clades corresponded to the speciation of SPG species, this led to the hypothesis that S6PDH and SOT are more relevant than SDH in the evolution of the sorbitol characteristic. The fact that only single SDH genes were identified in peach and mei (Fig. 2) supported this hypothesis. However, the surprising expansion of SDH genes in apple and pear caused a reconsideration of the evolution of SDH to the sorbitol characteristic, especially in apple and pear genomes. Although about half of the SDH in the pear genome were duplicated through WGD or segmental duplication (Fig. 2 and Table S3), such duplication type could not explain the expansion in the apple genome. If we take SDH class II [59] into account, we could reject the hypothesis that S6PDH and SOT are more relevant in the evolution of the sorbitol characteristic, because there were three peach and two mei genes in the SDH class II. Altogether, three gene families are important for the evolution of the sorbitol characteristic.
The evolution of plant traits may accompany selection pressure of related genes, either positive or purifying selection. We compared selection pressure in three gene families and confirmed the presence of strong purifying selection by codon bias analysis. Evolutionary rates are number of substitutions (fixed mutations) per unit of evolutionary time, which is typically estimated by the ratio ω = dN/dS (or Ka/Ks), where dN (Ks) indicates the non-synonymous substitution rate and dS (Ks) indicates the synonymous substitution rate [62]. The ratio ω is generally used as an indicator of positive selection (ω > 1) and purifying selection (ω < 1). Positive selection is the selection of beneficial alleles, while purifying selection is the selection against deleterious alleles [63]. The branch-specific model for the S6PDH, SDH and SOT gene families showed genes in SPG were found with evolutionary rates less than 1 (ω < 1), indicating purifying selection for all sorbitol metabolism pathway-related genes in this group. On the other hand, results for evolutionary rates for different branches with the multiple-ratio model indicated unbalanced evolutionary rates. The S6PDH clade A and SDH clade A were both found to be under strong purifying selection, while S6PDH clade B and SOT clade C both had with a relatively higher evolutionary rate, indicating reduced evolutionary constraints, which were driven by the result of relaxation of purifying selection [64]. This could be ascribed to the sorbitol character. Similarly, a higher evolutionary ratio was found to be associated with higher protein synthesis in dicots compared with monocots [30]. Although SDH Class II was not involved in this study, it was found to be positively selected [59]. Then, we estimated the codon bias to confirm the identified purifying selection. The results were confirmed by a relatively higher value of FOP in S6PDH and SDH genes in SPG. Different synonymous codons are favored by natural selection for translation efficiency and accuracy in different organisms [53]. Codon usage bias is not only a ubiquitous phenomenon observed in bacteria, plants and mammals [65, 66], but also plays a role as a means to fine-tune gene expression [67].
The duplicated members of sorbitol-related genes should lead to diversified gene expression patterns in the view of temporal and spatial differences. The expression analysis of S6PDH, SDH and SOT genes in the four SPG species supported the hypothesis that genes were differentially expressed in different tissues and different stages of fruit development.
Conclusion
In this study, we first performed comparative evolutionary analyses for three keys genes (S6PDH, SDH and SOT) involved in the sorbitol metabolism pathway in two groups of species, with (SPG) and without (SAG) sorbitol as the major photosynthetic product. We found that the number of genes in the three gene families were expanded in SPG through dispersed and tandem duplication. SDH and S6PDH clade A in SPG were found to be under strong purifying selection. Branch-site model analysis revealed SOT genes in SPG were under positive selection. Codon usage revealed a higher frequency of optimal codons for S6PDH and SDH than SOT genes, confirming the effect of purifying selection. Expression analyses in fruit and leaf tissues identified important genes involved in sorbitol metabolism. Overall, this study provides further insights for understanding the underlying molecular mechanism for sorbitol metabolism.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the NCBI repository.
SRR767660 in https://www.ncbi.nlm.nih.gov/sra/?term=SRR767660; SRR768128 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR768128; SRR768129 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR768129; SRR768130 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR768130; SRR768131 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR768131;SRR542478 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR542478; SRR542479 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR542479; SRR542480 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR542480; SRR542481 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR542481; SRR542482 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR542482;SRR654690 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR654690; SRR654692 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR654692; SRR654693 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR654693; SRR654695 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR654695; SRR654699 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR654699; SRR654700 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR654700;SRR531862 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR531862; SRR531863 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR531863; SRR531864 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR531864; SRR531865 in https://www.ncbi.nlm.nih.gov/sra/?term= SRR531865.
Not applicable.
References
Wang J, Nayak S, Koch K, Ming R. Carbon partitioning in sugarcane (Saccharum species). Front Plant Sci. 2013;4:201.
Shangguan L, Song C, Leng X, Kayesh E, Sun X, Fang J. Mining and comparison of the genes encoding the key enzymes involved in sugar biosynthesis in apple, grape, and sweet orange. Sci Hortic. 2014;165:311–8.
Wallaart RA. Distribution of sorbitol in Rosaceae. Phytochemistry. 1980;19(12):2603–10.
Webb KL, Burley JW. Sorbitol translocation in apple. Science. 1962;137(3532):766.
Potter D, Eriksson T, Evans RC, Oh S, Smedmark J, Morgan DR, et al. Phylogeny and classification of Rosaceae. Plant Syst Evol. 2007;266(1):5–43.
Ma C, Sun Z, Chen C, Zhang L, Zhu S. Simultaneous separation and determination of fructose, sorbitol, glucose and sucrose in fruits by HPLC-ELSD. Food Chem. 2014;145:784–8.
Li MJ, Li PM, Ma FW, Dandekar AM, Cheng LL. Sugar metabolism and accumulation in the fruit of transgenic apple trees with decreased sorbitol synthesis. Hortic Res. 2018;5:60.
Meng D, He MY, Bai Y, Xu HX, Dandekar AM, Fei ZJ, et al. Decreased sorbitol synthesis leads to abnormal stamen development and reduced pollen tube growth via an MYB transcription factor, MdMYB39L, in apple (Malus domestica). New Phytol. 2018;217(2):641–56.
Meng D, Li CL, Park HJ, Gonzalez J, Wang JY, Dandekar AM, et al. Sorbitol modulates resistance to Alternaria alternata by regulating the expression of an NLR resistance gene in apple. Plant Cell. 2018;30(7):1562–81.
Hirai M. Sorbitol-6-phosphate dehydrogenase from loquat fruit. Plant Physiol. 1979;63(4):715–7.
Zhou R, Cheng L, Wayne R. Purification and characterization of sorbitol-6-phosphate phosphatase from apple leaves. Plant Sci. 2003;165(1):227–32.
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–20.
Bantog NA, Yamada K, Niwa N, Shiratake K, Yamaki S. Gene expression of NAD(+)-dependent sorbitol dehydrogenase and NADP(+)-dependent sorbitol-6-phosphate dehydrogenase during development of loquat (Eriobotrya japonica Lindl.) fruit. J Japan Soc Hortic Sci. 2000;69(3):231–6.
Yamaki S. A sorbitol oxidase that converts sorbitol to glucose in apple leaf. Plant Cell Physiol. 1980;21(4):591–9.
Liang D, Cui M, Wu S, Ma FW. Genomic structure, sub-cellular localization, and promoter analysis of the gene encoding sorbitol-6-phosphate dehydrogenase from apple. Plant Mol Biol Report. 2012;30(4):904–14.
Zhang JY, Tian RR, Dong JL, Zhao K, Li TH, Wang T. Response and regulation of the S6PDH gene in apple leaves under osmotic stress. J Hortic Sci Biotechnol. 2011;86(6):563–8.
Gao JJ, Sun YR, Zhu B, Peng RH, Wang B, Wang LJ, et al. Ectopic expression of sorbitol-6-phosphate 2-dehydrogenase gene from Haloarcula marismortui enhances salt tolerance in transgenic Arabidopsis thaliana. Acta Physiol Plant. 2018;40(6):108.
Aguayo MF, Ampuero D, Mandujano P, Parada R, Munoz R, Gallart M, et al. Sorbitol dehydrogenase is a cytosolic protein required for sorbitol metabolism in Arabidopsis thaliana. Plant Sci. 2013;205-206:63–75.
Sola-Carvajal A, Garcia-Garcia MI, Garcia-Carmona F, Sanchez-Ferrer A. Insights into the evolution of sorbitol metabolism: phylogenetic analysis of SDR196C family. BMC Evol Biol. 2012;12:147.
Nosarzewski M, Downie AB, Wu BH, Archbold DD. The role of SORBITOL DEHYDROGENASE in Arabidopsis thaliana. Funct Plant Biol. 2012;39(6):462–70.
Almaghamsi A, Nosarzewski M, Kanayama Y, Archbold DD. Effects of abiotic stresses on sorbitol biosynthesis and metabolism in tomato (Solanum lycopersicum). Funct Plant Biol. 2021;48(3):286–97.
Shi XP, Ren JJ, Yu Q, Zhou SM, Ren QP, Kong LJ, et al. Overexpression of SDH confers tolerance to salt and osmotic stress, but decreases ABA sensitivity in Arabidopsis. Plant Biol. 2018;20(2):327–37.
Li F, Lei HJ, Zhao XJ, Tian RR, Li TH. Characterization of three sorbitol transporter genes in micropropagated apple plants grown under drought stress. Plant Mol Biol Report. 2012;30(1):123–30.
Gu C, Wu RF, Yu CY, Qi KJ, Wu C, Zhang HP, et al. Spatio-temporally expressed sorbitol transporters cooperatively regulate sorbitol accumulation in pear fruit. Plant Sci. 2021;303:110787.
Yu CY, Cheng HY, Cheng R, Qi KJ, Gu C, Zhang SL. Expression analysis of sorbitol transporters in pear tissues reveals that PbSOT6/20 is associated with sorbitol accumulation in pear fruits. Sci Hortic. 2019;243:595–601.
Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat Genet. 2010;42(10):833–9.
Wu J, Wang Z, Shi Z, Zhang S, Ming R, Zhu S, et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 2013;23(2):396–408.
Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 2013;45(5):487–94.
Zhang J. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18(6):292–8.
Li C, Li M, Dunwell JM, Zhang YM. Gene duplication and an accelerated evolutionary rate in 11S globulin genes are associated with higher protein synthesis in dicots as compared to monocots. BMC Evol Biol. 2012;12:15.
Li C, Li QG, Dunwell JM, Zhang YM. Divergent evolutionary pattern of starch biosynthetic pathway genes in grasses and dicots. Mol Biol Evol. 2012;29(10):3227–36.
Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28(1):33–6.
Koonin EV. Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. 2005;39:309–38.
Kristensen DM, Kannan L, Coleman MK, Wolf YI, Sorokin A, Koonin EV, et al. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics. 2010;26(12):1481–7.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402.
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39(Database issue):D225–9.
Loytynoja A, Goldman N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science. 2008;320(5883):1632–5.
Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27(8):1164–5.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25(7):1307–20.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42.
Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992;8(3):275–82.
Gelman A, Rubin DB. Inference from iterative simulation using multiple sequences. Stat Sci. 1992;7(4):457–72.
Sukumaran J, Holder MT. DendroPy: a Python library for phylogenetic computing. Bioinformatics. 2010;26(12):1569–71.
Huson DH, Scornavacca C. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol. 2012;61(6):1061–7.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49.
Wang D, Zhang Y, Zhang Z, Zhu J, Yu J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics. 2010;8(1):77–80.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91.
Guindon S, Rodrigo AG, Dyer KA, Huelsenbeck JP. Modeling the site-specific variation of selection patterns along lineages. P Natl Acad Sci USA. 2004;101(35):12957–62.
Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19(6):908–17.
Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87(1):23–9.
Novembre JA. Accounting for background nucleotide composition when measuring codon usage bias. Mol Biol Evol. 2002;19(8):1390–4.
Hershberg R, Petrov DA. General rules for optimal codon choice. Plos Genet. 2009;5(7):e1000556.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7(3):562–78.
Zhang Q, Chen W, Sun L, Zhao F, Huang B, Yang W, et al. The genome of Prunus mume. Nat Commun. 2012;3:1318.
Singh ND, Larracuente AM, Clark AG. Contrasting the efficacy of selection on the X and autosomes in Drosophila. Mol Biol Evol. 2008;25(2):454–67.
Singh ND, Koerich LB, Carvalho AB, Clark AG. Positive and purifying selection on the Drosophila Y chromosome. Mol Biol Evol. 2014;31(10):2612–23.
Jia Y, Wong DC, Sweetman C, Bruning JB, Ford CM. New insights into the evolutionary history of plant sorbitol dehydrogenase. BMC Plant Biol. 2015;15:101.
Li JM, Zheng DM, Li LT, Qiao X, Wei SW, Bai B, et al. Genome-wide function, evolutionary characterization and expression analysis of sugar transporter family genes in pear (Pyrus bretschneideri Rehd). Plant Cell Physiol. 2015;56(9):1721–37.
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36(Web Server issue):W5–9.
Echave J, Spielman SJ, Wilke CO. Causes of evolutionary rate variation among protein sites. Nat Rev Genet. 2016;17(2):109–21.
Morrell PL, Buckler ES, Ross-Ibarra J. Crop genomics: advances and applications. Nat Rev Genet. 2011;13(2):85–96.
Jacobsen MW, da Fonseca RR, Bernatchez L, Hansen MM. Comparative analysis of complete mitochondrial genomes suggests that relaxed purifying selection is driving high nonsynonymous evolutionary rate of the NADH2 gene in whitefish (Coregonus ssp.). Mol Phylogenet Evol. 2016;95:161–70.
Camiolo S, Melito S, Porceddu A. New insights into the interplay between codon bias determinants in plants. DNA Res. 2015;22(6):461–70.
Wang L, Roossinck MJ. Comparative analysis of expressed sequences reveals a conserved pattern of optimal codon usage in plants. Plant Mol Biol. 2006;61(4–5):699–710.
Quax TE, Claassens NJ, Soll D, van der Oost J. Codon bias as a means to fine-tune gene expression. Mol Cell. 2015;59(2):149–61.
Funding
This project was supported by the Jiangsu Agricultural Science and Technology Innovation Fund (CX (20)2020) and the seed industry promotion project of Jiangsu (JBGS (2021)022).
Author information
Authors and Affiliations
Contributions
LL analyzed the data and drafted the manuscripts. ML designed experiment and revised the manuscript. JW, HY and JMD revised the manuscript and provided guidance for article writing. JW and SZ supported for experimental design and revised the manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Query sequences of S6PDH, SDH and SOT. Supplementary Table 2. List of identified genes for S6PDH, SDH, and SOT gene families. Supplementary Table 3. Information of identified genes for S6PDH, SDH, and SOT gene families. Supplementary Table 4. Optimal codons for seven species.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, L., Li, M., Wu, J. et al. Genome-wide identification and comparative evolutionary analysis of sorbitol metabolism pathway genes in four Rosaceae species and three model plants. BMC Plant Biol 22, 341 (2022). https://doi.org/10.1186/s12870-022-03729-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03729-z