
Abstract
Background
Comparative genomic analysis exhibits dynamic evolution of plastid genome (plastome) in the clusioid clade of Malpighiales, which comprise five families, including multiple inversions and gene losses. Little is known about the plastome evolution in Hypericaceae, a large family in the clade. Only the plastome of one species, Cratoxylum cochinchinense, has been published.
Results
We generated a complete plastome sequence for Hypericum ascyron, providing the first complete plastome from the tribe Hypericeae (Hypericaceae). The H. ascyron plastome exhibits dynamic changes in gene and intron content, structure, and sequence divergence compared to the C. cochinchinense plastome from the tribe Cratoxyleae (Hypericaceae). Transcriptome data determined the evolutionary fate of the missing plastid genes infA, rps7, rps16, rpl23, and rpl32 in H. ascyron. Putative functional transfers of infA, rps7, and rpl32 were detected to the nucleus, whereas rps16 and rpl23 were substituted by nuclear-encoded homologs. The plastid rpl32 was integrated into the nuclear-encoded SODcp gene. Our findings suggested that the transferred rpl32 had undergone subfunctionalization by duplication rather than alternative splicing. The H. ascyron plastome rearrangements involved seven inversions, at least three inverted repeat (IR) boundary shifts, which generated gene relocations and duplications. Accelerated substitution rates of plastid genes were observed in the H. ascyron plastome compared with that of C. cochinchinense plastid genes. The higher substitution rates in the accD and clpP were correlated with structural change, including a large insertion of amino acids and losses of two introns, respectively. In addition, we found evidence of positive selection of the clpP, matK, and rps3 genes in the three branches related to H. ascyron. In particular, the matK gene was repeatedly under selection within the family Hypericaceae. Selective pressure in the H. ascyron matK gene was associated with the loss of trnK-UUU and relocation into the IR region.
Conclusions
The Hypericum ascyron plastome sequence provides valuable information for improving the understanding of plastome evolution among the clusioid of the Malpighiales. Evidence for intracellular gene transfer from the plastid to the nucleus was detected in the nuclear transcriptome, providing insight into the evolutionary fate of plastid genes in Hypericaceae.
Similar content being viewed by others


Background
Plastids are one of energy-producing eukaryotic organelles that originate from cyanobacterial-like endosymbionts. Plastids contain their own genome, which is highly reduced compared to the ancestral genome due to massive losses in genes or their transfers into the nuclear genome [1]. Moreover, gene transfer from plastids to the nucleus is an ongoing process [2, 3]. Among angiosperms, plastid genomes (plastomes) are highly conserved in structure, mainly encoding for the photosynthetic or transcription apparatus of the organelles and their own replication apparatus. The plastomes have a quadripartite structure with large and small single copy (LSC and SSC, respectively) regions separated by two inverted repeat (IR) regions, ranging from 120 to 170 kb in length [4]. However, several genome rearrangements have been found in Asteraceae [5], Campanulaceae [6], Fabaceae [7], Geraniaceae [8], Oleaceae [9], Papaveraceae [10], Plantaginaceae [11, 12], and Poaceae [13] across angiosperm plastomes. Previous studies have suggested that plastome rearrangements are correlated with the number and size of repeats [7, 8, 14]. IR expansions and contractions also contribute to plastome rearrangements and gene content variations [10, 14, 15]. Although gene and intron contents are generally conserved in the plastomes, comparative analyses of these contents in angiosperm plastomes showed a variation in the losses of 27 protein-encoding genes and 8 introns [16]. Among them, functional replacements of plastid genes by gene transfer to the nucleus or by nuclear homolog substitution have been documented for only 11 plastid genes: accD [17,18,19,20,21,22,23], infA [24, 25], rpl20 [26], rpl22 [26,27,28], rpl23 [29, 30], rpl32 [24, 25, 31,32,33], rps7 [26], rps15 [10], rps16 [25, 33,34,35], ycf1 and ycf2 [26].
The family Hypericaceae comprises of approximately 700 species in 9 genera, which belong to the clusioid of the Malpighiales [36]. Hypericaceae has been classified into three tribes: Cratoxyleae, Hypericeae, and Vismieae. The relationships between the genera within three tribes are unclear. Hypericum, a member of the tribe Hypericeae, is the largest genus in the family with more than 490 species and has a cosmopolitan distribution [37]. Some species in this genus are economically and medically important plants that produce different kinds of naphthodianthrones (especially hypericin), acylphloroglucinol derivatives, and flavonoid compounds [38], which are used as depression medications and painkillers [39, 40]. Despite the many therapeutic applications of Hypericum spp., a natural product genomics approach is limited. Only the one whole genome sequence data of H. perforatum have been reported [41], and those for complete plastid and mitochondrial genomes belonging to this genus are yet unreported.
In the order Malpighiales, which comprises of 16,000 species in 716 genera and 36 families, approximately 236 plastomes from 69 genera have been sequenced (National Center for Biotechnology Information; NCBI, accessed on June 11, 2021), and the average plastome size and GC content for the order were 155.6 kb and 36.5%, respectively. Previous studies of comparative analyses have revealed that the plastomes are highly conserved in structural organization with a few exceptions (Euphorbiaceae, Passifloraceae, and Podostemaceae) [42,43,44,45]. Sequencing data of the plastomes from this order have also provided excellent examples of gene and intron loss. The evolutionary fate of these missing plastid genes showed that they were functionally replaced by gene transfer to the nucleus or by gene substitution of a nuclear gene. For example, transfers of infA and rpl32 and substitution of rps16 are well-characterized examples in Malpighiales [26, 31, 32, 34, 46]. Recently, the evolutionary fate of plastid-encoded rps7, rpl20, rpoA, ycf1, and ycf2 have also been documented [26].
Although the Hypericaceae is one of the largest families of Malpighiales, complete plastome sequence has been reported for only one species, Cratoxylum cochinchinense, which belongs to the tribe Cratoxyleae (Hypericaceae); comparative genomic analysis for plastome evolution is very limited. In this study, we generated the complete plastome of H. ascyron, representing the first sequenced member of the tribe Hypericeae. The H. ascyron plastome organization is characterized, including functional replacements of five plastid genes to the nucleus. In addition, we analyzed H. acyron and nine published Malpighiales plastomes to examine patterns of plastome organization and highlighted nucleotide substitution rates within the clusioid of the Malpighiales.
Results
Plastome size and content of Hypericum ascyron
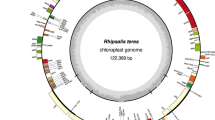
Illumina sequencing produced 32,049,446 PE raw reads, providing deep coverage (> 1,100 ×) for the H. ascyron plastome (Fig. 1A). The assembled plastome of H. ascyron exhibits a circular molecule with two copies of IRs of 26,846 bp, separating the LSC and SSC regions of 97,542 and 11,052 bp, respectively (Fig. 1B, Table S1). IR regions of H. ascyron are general in size relative to the other clusioid clade plastomes analyzed, however, the LSC is the largest and the SSC is the smallest SSC among them (Table S1). The largest number of repeat pairs (46) was found in H. ascyron within 10 Malpighiales plastomes analyzed (Table S1). The fewest repeat pairs (6) were found in the Sauvagesia rhodoleuca and Ctenolophon englerianus (Table S1). Hypericum ascyron contains a higher number of repeats, except a repeat smaller than 50 bp (Figure S1). Repeats larger than 500 bp were only present in H. ascyron (Figure S1).

Map of the plastid genome of Hypericum ascyron. A Graph showing the base per base depth of the sequencing coverage across the H. ascyron plastome with one inverted repeat (IR) region. B Genes on the inside and outside of each map are transcribed in the clockwise and counterclockwise directions, respectively. The thick lines on the plastid map indicate the inverted repeats (IRA and IRB) that separate the genome into large and small single-copy regions. Ψ denotes a pseudogene. Red arrows indicate expansion events. Colored arcs on the outside of the map correspond to the locally collinear blocks inferred by Mauve (see Fig. 4)
The H. ascyron plastome contained a set of genes encoding 74 proteins, 29 tRNAs and four rRNAs (Fig. 1, Table S1). The translation initiation factor A (infA), the ribosomal protein subunit S16 (rps16), and the tRNALys (trnK-UUU) genes were absent in the plastome. In addition, the ribosomal protein subunit S7 (rps7), L23 (rpl23), and L32 (rpl32) genes appeared to be pseudogenes due to frameshift indels or internal stop codons. The sequences of three pseudogenes were confirmed by Sanger sequencing. We found that the H. ascyron plastome had lost both introns in clpP, the cis-spliced second intron in rps12, the rpoC1 intron, and the second intron in ycf3.
The phylogenetic distribution of gene and intron content across the selected 10 Malpighiales plastomes showed that the events of gene and intron losses were species- and lineage-specific (Fig. 2). While all analyzed plastomes lacked the infA, the phylogenetic distribution of the rpl32 and rps16 indicated that the loss or pseudogenization of the two genes occurred independently in the selected Malphigales plastomes. All species in the clusioid lineage were missing the second intron of the ycf3 gene. Hypericum ascyron, C. cochinchinense, and Marathrum foeniculaceum shared the losses of rps16, the second clpP intron, and the cis-spliced second intron of rps12 and the pseudogenization of rps7. Among the Malphigales species analyzed, the absence of the trnK-UUU gene and the rpoC1 intron was unique to H. ascyron.

Phylogenetic distribution of gene/intron content (loss and gain) among the analyzed 10 Malpighiales
Identification of gene transfer and substitution
Our results showed that the H. ascyron plastome lacked five protein-coding genes, infA, rps7, rps16, rpl23, and rpl32. To identify the functional replacement of these genes to the nucleus, we performed “tblastn” searches against the H. ascyron transcriptome data using the amino acid sequences of each gene from Amborella trichopoda. Nuclear transcripts of only two genes, INFA and RPS7, were detected in the H. ascyron transcriptome (Fig. 3). Further, we found that predicted ORFs of two transcripts had extended amino acids upstream from the conserved domain, and that TargetP predicted a plastid transit peptide with high probability (INFA: 0.936 and RPS7: 0.769) (Fig. 3). To test the potential gene substitution by a nuclear homolog, we queried the amino acid sequences of the Medicago truncatula RPS16 and the spinach RPL23 against the H. ascyron transcriptome. The tblastn searches using nuclear the homologs of RPS16 and RPL23, two transcripts of each were identified (Figure S2A and B). Phylogenetic analysis of the nuclear-encoded RPS16 copies from H. ascyron and five other Malphigales with two Medicago copies suggested two different origins, although both were predicted to contain a mitochondrial transit peptide (Figure S2A). In regard to RPL23, only one transcript included a plastid transit peptide (0.919) (Figure S2B).

Amino acid sequence of nuclear-encoded plastid-targeted genes from Hypericum ascyron. Red boxes indicate the conserved domains, and pink boxes in the N-terminus indicate a transit peptide
To identify the nuclear-encoded RPL32, we queried the amino acid sequence of the SODcp-RPL32 chimeric gene from Passiflora contracta against the H. ascyron transcriptome. Two distinct transcripts were identified; one contains the conserved domain of “Cu–Zn Superoxide Dismutase superfamily” involving a plastid transit peptide (0.768) and the other contains the conserved domain of “ribosomal protein L32” and a plastid transit peptide (0.860) with a partial domain of “Cu–Zn Superoxide Dismutase superfamily” (Fig. 3). Amino acid sequence alignment of these two copies was divergent with low identity (23.9%) (Figure S2C). Compared with the available SODcp-RPL32 chimeric genes from Malpighiales species, the divergent pattern was similar to Populus trichocarpa (Figure S2C).
In addition, we also found the evidence of the functional replacement of these five genes to the nucleus in H. perforatum (Figure S3).
Structural evolution of the H. ascyron plastome
The H. ascyron plastome displayed dynamic changes, including multiple inversions, rearrangement, and IR boundary shifts, compared with the Mesua ferrea as an ancestral plastome structure (Fig. 4). Mauve alignment identified 16 locally collinear blocks (LCBs), suggesting seven inversions with 15 breakpoints: trnH-psbA, psbA-trnK, rps16-trnQ, trnE-trnT, trnT-psbD, trnT-trnL, ndhC-trnV, rbcL-accD, petA-psbJ, clpP-psbB, rpl2, ycf1, ycf1-ndhF, ndhH, and rps15-ycf1 (Fig. 4). Among them, one inversion in the H. ascyron IR region was associated with IR expansion (Figs. 1 and 4). Furthermore, we observed that nine regions (LCB 3, 4, 5, 6, 7, 8, 9, 10, and 14) were relocated because of genome rearrangements, including inversions (Figs. 1 and 4). Among them, a notable gene relocation was matK gene, which transferred from the LSC region into the IR region (Fig. 1). The loss of the trnK-UUU gene seems to be correlated with this event, leaving only 655 bp of the 5’ trnK intron with 73.1% identity (Figure S4A).

Structural alignments of plastomes from Hypericum ascyron with three related species using Mauve. The colored blocks represent collinear sequence blocks shared by all plastomes. Blocks drawn below the horizontal line indicate sequences found in an inverted orientation. Individual genes and strandedness are represented below each genome block. Only one copy of the inverted repeat (IR) is shown for each plastome and pink boxes below each plastome block indicate its IR
The structure of the H. ascyron plastome also has changed with IR boundary shifts in relation to the M. ferrea, generating multiple relocated genes (Fig. 4). For example, contraction (13 kb) at the LSC/IRA boundary excluded eight genes, from rpl2 to rps12, and two expansions at the IRB/SSC and IRA/SSC have resulted in the duplication of two genes, full copies of ndhF and ycf1 (Fig. 1).
We found that the partial sequence (371 bp) of the C-terminal region of rpl2 was located upstream of the rps19 gene (Figure S4B). The original copy of rpl2, downstream from rpl23, was truncated at the C-terminal (113 bp), but the conserved domain of ribosomal protein L2 was intact. The partial sequence was adjacent to the breakpoint and truncated original copy, suggesting the phenomenon was likely associated with the inferred inversion. In addition to the rpl2, the H. ascyron plastome contained four partial copies (513 bp) of surrounding the C-terminal portion of the accD (two are duplicated in the IR region) located in the IR region, with 96.7% identity (Figure S4C). The fragment next to the second accD copy is similar to a fragment located upstream of psbD gene, with 97.1% identity (Figure S4D).
In regard to an intact accD, its ORF in H. ascyron accD (3,204 bp) was highly expanded compared to the three related species (C. cochinchinense: 1,563 bp, M. foeniculaceum: 1,779 bp, and M. ferrea: 1,512 bp). The length of insertions in H. ascyron accD gene was confirmed by Sanger sequencing. Amino acid alignment of the four copies showed that C. cochinchinense, M. foeniculaceum, and H. ascyron contained amino acid insertions (Figure S5). Among them, we detected an interruption in the conserved domain of the H. ascyron accD gene, splitting the domain into two regions by a large insertion of 736 amino acids (Figure S5). The inserted regions in the H. ascyron accD gene was identified as multiple irregular amino acid repeats (Figure S6). However, the accD in C. cochinchinense and M. foeniculaceum contained only a small fraction of amino acids in the conserved domain (Figure S5).
The H. ascyron plastome contained a clpP-like ORF (1,074 bp), which was also expanded compared to the coding region of the M. ferrea clpP gene (591 bp). The nucleotide sequences of the clpP coding regions were highly divergent with low identity match between H. ascyron and the other three species, ranging from 36.3% to 42.7% (Figure S6). Amino acid alignment of the four copies showed that the H. ascyron clpP gene contained small insertions of amino acids at the N- and C-terminal regions (Figure S7).
Elevated substitution rates in the H. ascyron plastome
The nonsynonymous (dN) and synonymous (dS) substitution rates of the H. ascyron plastid genes were 4.8 times and 3.2 times significantly higher than that of the C. cochinchinense plastid genes, respectively (Wilcoxon rank-sum test, p < 0.0001; Figure S8). The H. ascyron exhibited elevated substitution rates in most of the individual plastid genes in comparison with that in C. cochinchinense, showing that the average substitution values in H. ascyron were 6.4 times higher for dN and 3.7 times higher for dS than that in C. cochinchinense (Fig. 5). In particular, the accD, clpP, rps3, rps11, and rps18 genes showed accelerated substitution rates compared with the C. cochinchinense (Fig. 5).

Variation in sequence divergence among Hypericum ascyron and Cratoxylum cochinchinense plastid protein-coding genes
Lineage-specific accelerations of dN and dS substitution rates were detected within the three species that experienced structural changes in accD and clpP (Fig. 6, Figures S7 and S9). By observing the three branches related to H. ascyron, we identified that the dN/dS values for the cemA, clpP, matK, psbN, rpl14, rps3, rps12, rps14, rps15, and rps18 genes were greater than one (Fig. 6). Likelihood ratio tests (LRTs) indicated that the branches, leading to H. ascyron/C. cochinchinense/M. foeniculaceum with regard to clpP, leading to H. ascyron/C. cochinchinense with regard to matK, and H. ascyron terminal with regard to matK and rps3, were significantly different (p < 0.05 after Bonferroni correction, Fig. 6), indicating positive selection.

Plastid sequence divergence among selected Malpighiales. In the ML tree on the left, genes were selected to span the ratios of nonsynonymous to synonymous substitution rates greater than one on the branches related to Hypericum ascyron. Phylograms (right) of concatenated genes depicting nonsynonymous (dN) and synonymous (dS) sequence divergence for three individual genes are shown. Genes that show positive selection are selected. Red branches indicate genes with significantly higher dN/dS values. All trees were drawn to the same scale
Discussion
In this study, we first generated the complete plastome of H. ascyron from the tribe Hypericeae (Hypericaceae). The size of H. ascyron plastome at 162,286 bp is close to the size of C. cochinchinense, which belongs to the tribe Cratoxyleae (Hypericaceae). However, the complete H. ascyron plastome exhibited extreme changes in gene and intron content, organization, and in the rate of sequence divergence compared with the other members of the same family. Our results provide insights for improving the understanding of plastome evolution among the family.
Evolutionary fate of the plastid gene losses in the H. ascyron plastome
The H. ascyron plastome lacked five protein-encoded genes (infA, rpl23, rpl32, rps7, and rps16), suggesting the activation of nuclear-encoded genes instead of the plastid genes. The phylogenetic distribution of the analyzed plastomes showed that the gene loss and pseudogenes are species- or lineage-specific (Fig. 2). These results suggest that functional replacement of plastid genes to the nucleus has occurred three times in the common ancestor of order Malpighiales (infA) or H. ascyron/C. cochinchinense/M. foeniculaceum (rps16 and rps7), and in H. ascyron (rpl23 and rpl32). Many studies have shown that plastid-encoded gene loss or pseudogenes in plastids occur after successful functional replacement to the nucleus. For example, intracellular gene transfer to the nucleus has been documented in angiosperms, including infA in Ranunculaceae [24, 25], rps7 in Passiflora [26], and rpl32 in Ranunculaceae [24, 25, 33], Rhizophoraceae and Salicaceae [31, 32]. With regard to rpl23 and rps16, gene substitution by the nuclear-encoded homologs have been reported: rpl23 in spinach [29] and Geranium [30], rps16 in Lupinus [35], Medicago and Populus [34], Passiflora [26], and Ranunculaceae [25, 33]. However, in Malpighiales, rpl32 is acquired by alternative splicing of the nuclear-encoded Cu–Zn superoxide dismutase (SOD) gene, which targets plastid [32]. This suggests that the chimeric gene occurred in the common ancestor of the order Malpighiales [31]. Our results also provide clear evidence for potential functional replacements of the five plastid genes by gene transfer to the nucleus (infA, rpl32, and rps7), and substitution (rpl23 and rps16) to the nucleus in H. ascyron (Fig. 3). All nuclear-encoded plastid-targeted INFA, RPS7, RPS16, RPL23, and RPL32 copies of H. ascyron contain transit peptides at N-terminal acquired by a novel, exon shuffling of an existing nuclear-encoded gene for plastids, and substituted by nuclear-encoded homologs. With regard to RPL32, two highly divergent copies suggested that H. ascyron underwent subfunctionalization by duplication rather than alternative splicing. Similar patterns that occur sub-functionalization are observed in Populus and Salix, whereas mangroves (Bruguiera gymnorrhiza) contain a chimeric gene via an alternative splicing [31, 32]. Shrestha et al. (2020) showed that the transit peptide of RPS7 was acquired by exon shuffling of a nuclear-encoded plastid-targeted thioredoxin m-type gene in Passiflora. However, the transit peptide acquisition of H. ascyron RPS7 is unclear from the present data. For a comprehensive understanding of the evolutionary fate of the five plastid genes within this family and the clusioid clade, additional nuclear genomic and transcriptomic data from other species will be required.
Structural variation in H. ascyron plastome
Multiple genomic rearrangements occurred in H. ascyron plastome, generating a number of gene relocations. Rearrangements promoted by homologous recombination between repeats has been proposed [47]. The number of repeat pairs in the H. ascyron plastome suggested that the repeats may facilitate rearrangements. Moreover, partial sequences of the accD throughout the LSC and IR regions can also rearrangements in H. ascyron plastome as repeated elements. In addition, the location of the matK as a free-standing gene, which was transferred into the IR region, suggested that genomic changes in H. ascyron involved multiple evolutionary mechanisms. Additional plastome sequences from other Hypericum species are needed to investigate the genome rearrangements including relocation of the matK.
IR boundary shifts (contraction and expansion) also affected the structure of the H. ascyron plastome, including gene reduction and duplication. For example, one copy of the rpl2, rpl23, trnI-CAU, ycf2, trnL-CAA, ndhB, rps7, and rps12 genes are completely lost, whereas the ndhF gene is completely duplicated. Two cases of duplication, matK and partial of accD, in the H. ascyron plastome are likely due to a combination of rearrangements and IR expansion. The IR contraction and expansion generated the longest LSC and the smallest SSC among the analyzed Malpighiales, although IR size was median size (Table S1). The IR boundary shifts have occurred multiple times across angiosperm plastomes, which is caused by double-strand break model [48, 49].
Substitution rate variation in H. ascyron plastome
A gene-specific rate increase compared with that in the C. cochinchinense plastid genes was observed in H. ascyron plastid genes (Fig. 5). Previous studies have shown a positive correlation between substitution rates and genomic rearrangements [7, 8, 14]. Multiple genomic changes including inversions in the H. ascyron can influence the elevated substitution rates in the plastid genes (Fig. 4). In addition, a positive effect of structural evolution on substitution rates of the H. ascyron accD and clpP genes also were identified. Highly divergent, intronless clpP gene in H. ascyron plastome was detected with greatly higher dN and dS values compared with the analyzed species. Likewise, correlation between elevated substitution rates and the loss of introns of the clpP gene has been observed in some angiosperm lineages, Geranium [22], legume [50], and Silene [51]. Possible mechanisms of intron loss have been proposed: direct genomic deletion, exonization of intron, and retroprocessing [52]. The phylogenetic distribution showed that the loss of the second intron occurred in the common ancestor of H. ascyron, C. cochinchinense and M. foeniculaceum followed by loss of the first intron independently in H. ascyron (Fig. 2), suggesting that different mechanisms may have been activated for each intron loss. In particular, our analysis showed evidence of positive selection on the branch leading to H. ascyron/C. cochinchinense/M. foeniculaceum (Fig. 6). In the plastid accD, the insertion of amino acids is also correlated with the accelerated substitution rates (Figure S9). Interruption of the accD was identified in Geranium [22] and Lamprocapnos [10] and the genes exhibit elevated substitution rates, although the patterns of insertion are different. However, the force driving the positive selection on the H. ascyron terminal branch with regard to rps3 is unclear.
In the plant, maturase K (matK) is located within the intron of trnK-UUU in the plastome. This matK product is essential for the splicing of group II introns, including cis-spliced introns of atpF, rpl2, rps12, trnV-UVC, trnI-GAU, trnA-UGC, and trnK-UUU. The loss of the trnK-UUU gene has been found in Cuscuta [53], Epifagus [54], Taxillus [55] and Erodium [14]. In H. ascyron, the surrounding regions of the trnK-UUU exons were lost and the matK was retained as a free-standing gene in the plastome. The H. ascyron plastome contained the five introns, although the cis-spliced rps12 and trnK-UUU introns were lost, indicating the functional necessity of the matK. The dN/dS values of the branches, leading to H. ascyron/C. cochinchinense and H. ascyron terminal were greater than one, and the LRT suggested that the matK gene underwent positive selection. The forces driving the positive selection in the H. ascyron matK are likely associated with the loss of trnK-UUU and relocation. However, C. cochinchinense matK is encoded within the intron of trnK-UUU in its plastome and does not relocate. The plastid matK in the lineage leading to H. ascyron/C. cochinchinense is likely to experience altered selection pressures. A complete understanding of these patterns of higher substitution rates requires sufficient samples from this family.
Conclusions
The plastome sequence of H. ascyron, a member of the tribe Hypericeae, provides new insights into the plastome evolution within Hypericiaceae. The plastome exhibits a number of unusual phenomena, including genome rearrangements, gene and intron losses, and elevated substitution rates compared with the C. cochinchinense plastome. Nuclear transcriptome data provide clear evidence for functional replacements by gene transfers (infA, rps7, and rpl32) or substitution (rps16 and rpl23) from the plastids to the nucleus.
Methods
DNA isolation, sequencing, assembly, and annotation
Hypericum ascyron was collected from Mt. Cheayak, Yeongcheon-si, Gyeongsangbuk-do, South Korea. The plant material used in this study was obtained from the wild and a voucher specimen was deposited in the Yeungnam University Herbarium (YNUH0202519 identified by SeonJoo Park). Experimental study on the plant, including collection of the material, comply with institutional, national, and international guidelines. Total DNA was isolated from fresh leaves (100 mg) using the DNeasy Plant Mini Kit (Qiagen Inc., GmbH, Germany). Total DNA was processed for genomic library preparation using the Hiseq2500 platform (Illumina, San Diego, CA, USA), generating 5.6 Gb of 2 × 150 bp paired-end (PE) reads from a 550 bp library. PE reads were used for de novo assembly using Velvet v1.2.10 assembler [56] with multiple k-mers ranging from 99 to 145 and expected coverage values (100, 200, 300, 400, and 500). To evaluate the depth of coverage, PE reads were mapped to the plastome with one IR region using Bowtie2 v2.2.6 [57]. The plastome was annotated using Geneious R11 v11.0.5 (https://www.geneious.com) with the protein coding genes of Nicotiana tabacum as a reference, and the open reading frame (ORF) of the plastome was evaluated. All tRNA genes were identified using tRNAscan-SE v2.0.3 [58] and ARAGON v1.2.38 [59]. The OGDraw v1.3.1 [60] was used to draw the circular plastome of H. ascyron. The annotated plastome sequence was deposited in GenBank (accession number MZ424306).
To check the assembly errors in the accD, rpl23, rpl32, rps7 copies, polymerase chain reaction (PCR) was carried out with specific primer pairs (Table S2), using methods described previously [10]. The PCR products were purified using the Solg™ Gel & PCR extraction system (Solgent Co., Daejeon, South Korea) following the manufacturer’s protocol and sequenced using an ABI 3730xl DNA Analyzer (Applied Biosystems, California, USA) at Solgent Co. Repeats in the accD gene were identified using “Find Repeats” option in Geneious R11 with minimum repeat length of 20 bp.
Validation of functional replacements of plastid genes to the nucleus
Total RNA was extracted from fresh leaves of H. ascyron using the HiGene™ Total RNA Prep Kit (ver. 2.0) (Biofact, Daejeon, South Korea) following the manufacture’s protocol. The RNA was sequenced using the Illumina HiSeq4000 platform with 2 × 150 bp PE reads. Error correction was performed using Rcorrector [61] with default parameters. To identify nuclear-encoded plastid-targeted genes, the transcriptome from H. ascyron was assembled using Trinity v2.11.0 [62] with the trimmomatic flag. Potential transcripts were identified using “tblastn” searches of the plastid-encoded genes (infA, rps7, rps16, rpl23, and rpl32 from Amborella trichopoda plastome; NC_005086) and nuclear-encoded genes (RPS16 from Medicago truncatula; AB365526, RPL23 from Spinacia oleracea; Q9LWB5, SODcp-RPL32 from Passiflora contracta; MT259558) against the H. ascyron transcriptome. The open reading frames (ORFs) in the transcripts were determined and translated using Geneious R11 and subsequently were used as queries. TargetP v.1.1 [63] was used to predict transit peptides, and the NCBI Conserved Domain Database (CDD) was used to annotate the functional domain of proteins [64]. The published PE reads of Hypericum perforatum (SRR8438984) was also used. Transcriptome assembly and identification of the nuclear-encoded plastid-targeted genes were performed as described above.
Comparative analyses
Plastome rearrangements in H. ascyron were compared with nine genera from nine families (Bonnetiaceae, Calophylleae, Clusiaceae, Ctenolophonaceae, Erythroxylaceae, Hypericaceae, Ochnaceae, Podostemoideae, and Rhizophoraceae) of Malphigales (Table S1) using Mauve v2.3.1 [65] in Geneious R11 with default parameters. Repetitive sequences were identified by performing “blastn” searches [66] of each plastome against itself with an e-value cutoff of 1e-10 after removing one copy of IR.
Phylogenetic and substitution rate estimation
To reconstruct phylogenetic relationships among other Malphigales, 70 plastid protein-coding genes from the selected 10 plastomes were extracted (Table S3). The individual genes were aligned using the Translation Align-Maximum likelihood (ML)-based MAFFT v7.450 [67] in Geneious R11 and concatenated to a single alignment data set. The maximum likelihood (ML) tree was generated using IQ-TREE v1.6.2 [68] with an ultrafast bootstrap algorithm (1000 replicates). Nonsynonymous (dN) and synonymous (dS) nucleotide substitutions for all selected gene trees were calculated in PAML v4.8 [69] using the CODEML program with the F3 × 4 codon frequency mode. To test for branch sites under positive selection, we used the “adaptive BSREL” model [70] implemented in HyPhy v2.5 [71] using the Datamonkey server [72], which performs a series of likelihood ratio tests (LRTs) using the Holm-Bonferroni correction.
Availability of data and materials
The data sets supporting the results of this article are included in additional files. Complete plastid genome and gene sequences are available in the GenBank (https://www.ncbi.nlm.nih.gov/nuccore/MZ424306, OM967034-OM967041).
Abbreviations
- d N :
-
Number of substitutions per nonsynonymous site
- d S :
-
Number of substitutions per synonymous site
- LRT:
-
Likelihood ratio test
- ORF:
-
Open reading frame
- LSC:
-
Large single copy
- SSC:
-
Small single copy
- IR:
-
Inverted repeat
References
Timmis JN, Ayliffe MA, Huang CY, Martin W. Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat Rev Genet. 2004;5(2):123–35.
Huang CY, Ayliffe MA, Timmis JN. Direct measurement of the transfer rate of chloroplast DNA into the nucleus. Nature. 2003;422(6927):72–6.
Stegemann S, Hartmann S, Ruf S, Bock R. High-frequency gene transfer from the chloroplast genome to the nucleus. Proc Natl Acad Sci USA. 2003;100(15):8828–33.
Ruhlman TA, Jansen RK. Plastid genomes of flowering plants: essential principles. In: Maliga P, editor. Chloroplast Biotechnology: Methods and Protocols. New York: Springer US; 2021. p. 3–47.
Kim K-J, Choi K-S, Jansen RK. Two chloroplast DNA inversions originated simultaneously during the early evolution of the sunflower family (Asteraceae). Mol Biol Evol. 2005;22(9):1783–92.
Cosner ME, Raubeson LA, Jansen RK. Chloroplast DNA rearrangements in Campanulaceae: phylogenetic utility of highly rearranged genomes. BMC Evol Biol. 2004;4(1):1–17.
Cai Z, Guisinger M, Kim H-G, Ruck E, Blazier JC, McMurtry V, Kuehl JV, Boore J, Jansen RK. Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. J Mol Evol. 2008;67(6):696–704.
Weng M-L, Blazier JC, Govindu M, Jansen RK. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol Biol Evol. 2014;31(3):645–59.
Lee H-L, Jansen RK, Chumley TW, Kim K-J. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol Biol Evol. 2007;24(5):1161–80.
Park S, An B, Park S. Reconfiguration of the plastid genome in Lamprocapnos spectabilis: IR boundary shifting, inversion, and intraspecific variation. Sci Rep. 2018;8(1):13568.
Zhu A, Guo W, Gupta S, Fan W, Mower JP. Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016;209(4):1747–56.
Mower JP, Guo W, Partha R, Fan W, Levsen N, Wolff K, Nugent JM, Pabón-Mora N, González F. Plastomes from tribe Plantagineae (Plantaginaceae) reveal infrageneric structural synapormorphies and localized hypermutation for Plantago and functional loss of ndh genes from Littorella. Mol Phylogenet Evol. 2021;162:107217.
Doyle JJ, Davis JI, Soreng RJ, Garvin D, Anderson MJ. Chloroplast DNA inversions and the origin of the grass family (Poaceae). Proc Natl Acad Sci USA. 1992;89(16):7722–6.
Guisinger MM, Kuehl JV, Boore JL, Jansen RK. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol Biol Evol. 2011;28(1):583–600.
Weng ML, Ruhlman TA, Jansen RK. Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 2017;214(2):842–51.
Jansen RK, Cai Z, Raubeson LA, Daniell H, dePamphilis CW, Leebens-Mack J, Müller KF, Guisinger-Bellian M, Haberle RC, Hansen AK. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci USA. 2007;104(49):19369–74.
Konishi T, Shinohara K, Yamada K, Sasaki Y. Acetyl-CoA Carboxylase in higher plants: most plants other than gramineae have both the prokaryotic and the eukaryotic forms of this enzyme. Plant Cell Physiol. 1996;37(2):117–22.
Gornicki P, Faris J, King I, Podkowinski J, Gill B, Haselkorn R. Plastid-localized acetyl-CoA carboxylase of bread wheat is encoded by a single gene on each of the three ancestral chromosome sets. Proc Natl Acad Sci USA. 1997;94(25):14179–84.
Schulte W, Töpfer R, Stracke R, Schell J, Martini N. Multi-functional acetyl-CoA carboxylase from Brassica napus is encoded by a multi-gene family: Indication for plastidic localization of at least one isoform. Proc Natl Acad Sci USA. 1997;94(7):3465–70.
Magee AM, Aspinall S, Rice DW, Cusack BP, Sémon M, Perry AS, Stefanović S, Milbourne D, Barth S, Palmer JD. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010;20(12):1700–10.
Babiychuk E, Vandepoele K, Wissing J, Garcia-Diaz M, De Rycke R, Akbari H, Joubès J, Beeckman T, Jänsch L, Frentzen M, et al. Plastid gene expression and plant development require a plastidic protein of the mitochondrial transcription termination factor family. Proc Natl Acad Sci USA. 2011;108(16):6674–9.
Park S, Ruhlman TA, Weng M-L, Hajrah NH, Sabir JSM, Jansen RK. Contrasting patterns of nucleotide substitution rates provide insight into dynamic evolution of plastid and mitochondrial genomes of Geranium. Genome Biol Evol. 2017;9(6):1766–80.
Sabir J, Schwarz E, Ellison N, Zhang J, Baeshen NA, Mutwakil M, Jansen R, Ruhlman T. Evolutionary and biotechnology implications of plastid genome variation in the inverted-repeat-lacking clade of legumes. Plant Biotechnol J. 2014;12(6):743–54.
Park S, Jansen RK, Park S. Complete plastome sequence of Thalictrum coreanum (Ranunculaceae) and transfer of the rpl32 gene to the nucleus in the ancestor of the subfamily Thalictroideae. BMC Plant Biol. 2015;15(1):40.
Park S, Park S. Large-scale phylogenomics reveals ancient introgression in Asian Hepatica and new insights into the origin of the insular endemic Hepatica maxima. Sci Rep. 2020;10(1):16288.
Shrestha B, Gilbert LE, Ruhlman TA, Jansen RK. Rampant Nuclear Transfer and Substitutions of Plastid Genes in Passiflora. Genome Biol Evol. 2020;12(8):1313–29.
Gantt JS, Baldauf SL, Calie PJ, Weeden NF, Palmer JD. Transfer of rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. EMBO J. 1991;10(10):3073–8.
Jansen RK, Saski C, Lee S-B, Hansen AK, Daniell H. Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Mol Biol Evol. 2011;28(1):835–47.
Bubunenko M, Schmidt J, Subramanian A. Protein substitution in chloroplast ribosome evolution: a eukaryotic cytosolic protein has replaced its organelle homologue (L23) in spinach. J Mol Biol. 1994;240(1):28–41.
Weng M-L, Ruhlman TA, Jansen RK. Plastid-nuclear interaction and accelerated coevolution in plastid ribosomal genes in Geraniaceae. Genome Biol Evol. 2016;8(6):1824–38.
Cusack BP, Wolfe KH. When gene marriages don’t work out: divorce by subfunctionalization. Trends Genet. 2007;23(6):270–2.
Ueda M, Fujimoto M, Arimura S-i, Murata J, Tsutsumi N, Kadowaki K-i. Loss of the rpl32 gene from the chloroplast genome and subsequent acquisition of a preexisting transit peptide within the nuclear gene in Populus. Gene. 2007;402(1–2):51–6.
Park S, An B, Park S. Recurrent gene duplication in the angiosperm tribe Delphinieae (Ranunculaceae) inferred from intracellular gene transfer events and heteroplasmic mutations in the plastid matK gene. Sci Rep. 2020;10(1):2720.
Ueda M, Nishikawa T, Fujimoto M, Takanashi H, Arimura S-i, Tsutsumi N, Kadowaki K-i. Substitution of the gene for chloroplast RPS16 was assisted by generation of a dual targeting signal. Mol Biol Evol. 2008;25(8):1566–75.
Keller J, Rousseau-Gueutin M, Martin GE, Morice J, Boutte J, Coissac E, Ourari M, Aïnouche M, Salmon A, Cabello-Hurtado F, et al. The evolutionary fate of the chloroplast and nuclear rps16 genes as revealed through the sequencing and comparative analyses of four novel legume chloroplast genomes from Lupinus. DNA Res. 2017;24(4):343–58.
Wurdack KJ, Davis CC. Malpighiales phylogenetics: Gaining ground on one of the most recalcitrant clades in the angiosperm tree of life. Am J Bot. 2009;96(8):1551–70.
Stevens PF. Hypericaceae. In: Flowering Plants · Eudicots: Berberidopsidales, Buxales, Crossosomatales, Fabales pp, Geraniales, Gunnerales, Myrtales pp, Proteales, Saxifragales, Vitales, Zygophyllales, Clusiaceae Alliance, Passifloraceae Alliance, Dilleniaceae, Huaceae, Picramniaceae, Sabiaceae. Edited by Kubitzki K. Berlin, Heidelberg: Springer Berlin Heidelberg; 2007: 194-201
Mat A. An overview on Hypericum species of Turkey. J Pharmacogn Phytotherapy. 2013;5(3):38–46.
Lieberman S. Nutriceutical review of St. John’s wort (Hypericum perforatum) for the treatment of depression. J Womens Health. 1998;7(2):177–82.
Barnes J, Anderson LA, Phillipson JD. St John’s wort (Hypericum perforatum L.): a review of its chemistry, pharmacology and clinical properties. J Pharm Pharmacol. 2010;53(5):583–600.
Zhou W, Wang Y, Li B, Petijová L, Hu S, Zhang Q, Niu J, Wang D, Wang S, Dong Y, et al. Whole-genome sequence data of Hypericum perforatum and functional characterization of melatonin biosynthesis by N-acetylserotonin O-methyltransferase. J Pineal Res. 2021;70(2):e12709.
Tangphatsornruang S, Uthaipaisanwong P, Sangsrakru D, Chanprasert J, Yoocha T, Jomchai N, Tragoonrung S. Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene. 2011;475(2):104–12.
Bedoya AM, Ruhfel BR, Philbrick CT, Madriñán S, Bove CP, Mesterházy A, Olmstead RG. Plastid genomes of five species of Riverweeds (Podostemaceae): structural organization and comparative analysis in malpighiales. Front Plant Sci. 2019;10:1035.
Cauz-Santos LA, da Costa ZP, Callot C, Cauet S, Zucchi MI, Bergès H, van den Berg C, Vieira MLC. A repertory of rearrangements and the loss of an inverted repeat region in passiflora chloroplast genomes. Genome Biol Evol. 2020;12(10):1841–57.
Jin D-M, Jin J-J, Yi T-S. Plastome structural conservation and evolution in the clusioid clade of malpighiales. Sci Rep. 2020;10(1):9091.
Alqahtani AA, Jansen RK. The evolutionary fate of rpl32 and rps16 losses in the Euphorbia schimperi (Euphorbiaceae) plastome. Sci Rep. 2021;11(1):7466.
Wicke S, Schneeweiss GM, dePamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3):273–97.
Goulding SE, Wolfe K, Olmstead R, Morden C. Ebb and flow of the chloroplast inverted repeat. Mol Gen Genet MGG. 1996;252(1):195–206.
Wang R-J, Cheng C-L, Chang C-C, Wu C-L, Su T-M, Chaw S-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol Biol. 2008;8(1):1–14.
Dugas DV, Hernandez D, Koenen EJ, Schwarz E, Straub S, Hughes CE, Jansen RK, Nageswara-Rao M, Staats M, Trujillo JT. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions and accelerated rate of evolution in clpP. Sci Rep. 2015;5(1):1–13.
Erixon P, Oxelman B. Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the chloroplast clpP1 gene. PLoS One. 2008;3(1):e1386.
Hepburn NJ, Schmidt DW, Mower JP. Loss of two introns from the Magnolia tripetala Mitochondrial cox2 gene implicates horizontal gene transfer and gene conversion as a novel mechanism of intron loss. Mol Biol Evol. 2012;29(10):3111–20.
McNeal JR, Kuehl JV, Boore JL, Leebens-Mack J, Depamphilis CW. Parallel loss of plastid introns and their maturase in the genus Cuscuta. PLoS One. 2009;4(6):e5982.
Ems SC, Morden CW, Dixon CK, Wolfe KH, de Pamphilis CW, Palmer JD. Transcription, splicing and editing of plastid RNAs in the nonphotosynthetic plant Epifagus virginiana. Plant Mol Biol. 1995;29(4):721–33.
Li Y, Zhou J-g, Chen X-l, Cui Y-x, Xu Z-c, Li Y-h, Song J-y, Duan B-z, Yao H. Gene losses and partial deletion of small single-copy regions of the chloroplast genomes of two hemiparasitic Taxillus species. Sci Rep. 2017;7(1):12834.
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–9.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9.
Chan PP, Lowe TM. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In: Kollmar M, editor. Gene Prediction: Methods and Protocols. New York: Springer New York; 2019. p. 1–14.
Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32(1):11–6.
Greiner S, Lehwark P, Bock R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019;47(W1):W59–64.
Song L, Florea L. Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. GigaScience. 2015;4(1):48.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP SignalP and related tools. Nat Protoc. 2007;2(4):953–71.
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2010;39(suppl_1):D225–9.
Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–403.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10(1):421.
Katoh K, Misawa K. Kuma Ki, Miyata T: MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–66.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2014;32(1):268–74.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91.
Smith MD, Wertheim JO, Weaver S, Murrell B, Scheffler K, Kosakovsky Pond SL. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol. 2015;32(5):1342–53.
Kosakovsky Pond SL, Poon AFY, Velazquez R, Weaver S, Hepler NL, Murrell B, Shank SD, Magalis BR, Bouvier D, Nekrutenko A, et al. HyPhy 2.5—A customizable platform for evolutionary hypothesis testing using phylogenies. Mol Biol Evol. 2019;37(1):295–9.
Weaver S, Shank SD, Spielman SJ, Li M, Muse SV, Kosakovsky Pond SL. Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol Biol Evol. 2018;35(3):773–7.
Acknowledgements
Not applicable.
Funding
This work was supported by the 2021 Yeungnam Universtiy Grant (221A061009), South Korea.
Author information
Authors and Affiliations
Contributions
JCS performed the experiments, generated datasets, and wrote the first draft of the manuscript. SP contributed to the design of the project, assembled, performed analyses, prepared figures, and read/edited the manuscript. SJP contributed to project design and read/edited the manuscript. All authors read and approved the final draft of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Experimental study on the plant, including collection of the material, comply with institutional, national, and international guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Claude, SJ., Park, S. & Park, S. Gene loss, genome rearrangement, and accelerated substitution rates in plastid genome of Hypericum ascyron (Hypericaceae). BMC Plant Biol 22, 135 (2022). https://doi.org/10.1186/s12870-022-03515-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03515-x