
Abstract
Background
The East Asian subtropical evergreen broad-leaved forests (EBLFs) harbor remarkable biodiversity. However, their historical assembly remains unclear. To gain new insights into the assembly of this biome, we generated a molecular phylogeny of one of its essential plant groups, the tribe Perseeae (Lauraceae).
Results
Our plastid tree topologies were robust to analyses based on different plastid regions and different strategies for data partitioning, nucleotide substitution saturation, and gap handling. We found that tribe Perseeae comprised six major clades and began to colonize the subtropical EBLFs of East Asia in the early Miocene. The diversification rates of tribe Perseeae accelerated twice in the late Miocene.
Conclusions
Our findings suggest that the intensified precipitation in East Asia in the early Miocene may have facilitated range expansions of the subtropical EBLFs and establishment of tribe Perseeae within this biome. By the late Miocene, species assembly and diversification within the EBLFs had become rapid.
Similar content being viewed by others

Background
Subtropical evergreen broad-leaved forests (EBLFs) are an important vegetation type, and are primarily distributed in East Asia [1]. EBLFs are characterized by species that depend on the regional monsoons, including Castanopsis Spach, Lithocarpus Bl., Quercus L. sect. Cyclobalanopsis Schneid. (Fagaceae), Machilus Nees (Lauraceae), and Schima Reinw. (Theaceae) [2]. Subtropical EBLFs presently occur between 24°N and 32°N and 99°E and 123°E in China, and they once covered about 25% of the total area of the country [3]. Unfortunately, over the past decades, the subtropical EBLFs have become dramatically reduced because of human activities [4].
Biomes, such as the EBLFs of East Asia, are assembled via a complex network of processes that occur at many spatial and temporal scales [5]. Assembly is accomplished from a regional species pool and is constrained by the unique biology of the species, their ecological tolerances, and their evolutionary histories [6]. Thus, assembly of a biome may be best understood through integrating over local, short-term ecological processes and longer-term, broader-scale evolutionary ones. Achieving a better understanding of the assembly of a biome and its historical dynamics can yield insights into the future of its biodiversity in a changing world [7, 8]. However, our knowledge regarding the historical assembly of the East Asian subtropical EBLFs remains limited.
Assembly and historical dynamics of regional biomes can be inferred by reconstructing ancestral states across dated phylogenies. In the past decades, several approaches to performing such analyses have been proposed (e.g., [9,10,11]). These approaches have been widely used to investigate the ancestral state of taxa and the evolution of biotas. For example, De-Nova et al. [12] employed a maximum likelihood approach to infer that seasonally dry tropical forest was the ancestral habitat of Bursera Jacq. (Burseraceae). The subtropical EBLFs of East Asia have also been investigated using ancestral state reconstructions (ASRs) of plant lineages. For example, Yu et al. [13] reconstructed the ancestral habitat of Theaceae and found that this family colonized the East Asian subtropical EBLFs in the early Miocene. Their results suggested that the evolution of the subtropical EBLFs of East Asia was facilitated by two intensifications of the East Asian monsoon (EAM), one at the Oligocene-Miocene (O-M) boundary and the other in the late Miocene [13]. In addition to ancestral range/state reconstructions, diversification rate analyses have also been used to explore the historical dynamics of the subtropical EBLFs of East Asia. For example, Yu et al. [13] and Wang et al. [14] detected accelerated diversification rates in Theaceae and Lardizabalaceae, respectively, in the Miocene.
Within the East Asian subtropical EBLFs, keystone families primarily comprise the Fagaceae, Lauraceae, Theaceae, and Magnoliaceae [15], all of which consist largely or entirely of tree species. Studies based on these keystone lineages may yield the most robust and accurate insights into the development of the EBLFs. Nevertheless, many prior studies employing historical biogeographic or diversification rate analyses have focused on herbs (Coptis Salisb.) [16], woody climbers, or shrubs (Sabia Colelbr. Lardizabalaceae) [14, 17]. Only two keystone plant groups of the East Asian subtropical EBLFs, Theaceae [13] and Quercus sect. Cyclobalanopsis (Fagaceae) [8], have been studied to investigate the historical assembly of this biome. The evolutionary histories of the other keystone plant groups within the EBLFs and their roles as components of biome assembly remain unclear.
In this study, we investigated historical assembly and change in the subtropical EBLFs of East Asia using the tribe Perseeae in the keystone family Lauraceae. Tribe Perseeae comprises eight genera and ca. 385 species (Table S1) that are shrubs and trees of great economic and ecological importance [18]. Species of tribe Perseeae are mainly distributed in tropical and subtropical Asia and belong to Alseodaphne Nees, Alseodaphnopsis H.W. Li & J. Li, Apollonias Nees, Dehaasia Bl., Machilus Nees, Nothaphoebe Bl., Persea Mill., and Phoebe Nees [19,20,21]. Among these genera, species of Machilus and Phoebe are particularly abundant in subtropical EBLFs of East Asia [15]. In Machilus, 40 of ca. 114 (35.1%) species are endemic to the East Asian subtropical EBLFs, and 12 of ca. 51 (23.5%) Phoebe species are endemic in this biome [18, 22]. An additional 33 species of Machilus (28.9%) occur more broadly within both the tropical forests and subtropical EBLFs of East Asia, and the same is true for 11 species of Phoebe (21.6%). Overall, the distributional pattern of tribe Perseeae and its abundance within the subtropical EBLFs of East Asia as a member of an ecologically critical family make it an ideal system for investigating the assembly and historical dynamics of this biome.
Although tribe Perseeae is an essential group within the East Asian subtropical EBLFs, its phylogenetic relationships are not fully resolved, partly due to hybridization among species and taxonomic difficulties [23, 24]. For example, a molecular phylogeny by Li et al. [21] involved considerable sampling at the species level, but the major clades of tribe Perseeae were not strongly supported based on the ITS and intron II of LEAFY. More recently, well-resolved phylogenies of tribe Perseeae were reconstructed using plastid genomes (plastomes), but Machilus and Phoebe were poorly sampled in these studies [25, 26].
In this study, we sampled 52 species of tribe Perseeae, especially within the genera Machilus and Phoebe, to reconstruct a robust phylogeny of the tribe based on plastomes. Our sampling covered all major clades present in previous studies [21, 25, 26] and the key distributional ranges in Asia and the Americas, as well as seven of eight genera (excluding Apollonias, which is monotypic and distributed in India). We then carried out molecular dating analyses, estimations of diversification rates, and ASR to understand the evolutionary dynamics of tribe Perseeae, especially within the subtropical EBLFs of East Asia. Based on these results, we used tribe Perseeae as a proxy to infer aspects of assembly and historical change in the East Asian subtropical EBLFs. Thus, our work provides an additional evolutionary perspective on the historical dynamics of EBLFs in subtropical Asia based on a plant lineage that is dominant within the biome.
Results
Plastome features
Forty-four complete plastomes were newly sequenced and assembled, with lengths ranging from 151,780 bp (Sassafras tzumu Hemsl.) to 152,920 bp (Alseodaphnopsis sichourensis H. W. Li & J. Li) and AT contents ranging from 60.8 to 60.9%. All plastomes shared a typical quadripartite structure (Fig. S1): a large single-copy region (LSC), a small single-copy region (SSC), and two copies of an inverted repeat region (IR). Each plastome contained 113 distinct genes, including 30 transfer RNAs (tRNAs), four ribosomal RNAs (rRNAs), and 79 protein-coding genes. Among the 113 genes, 18 contained introns.
We extracted 242 loci from 61 complete plastomes and aligned them prior to phylogenetic analyses. Among these loci, ycf1, ycf2, and ndhF-rpl32 contained more variable sites and parsimony informative sites than other loci (Table S2). The 242 loci had a large range of AT contents, from 36.5% (trnD-GUC) to 90.5% (rpl2-rpl23), indicating an uneven distribution of AT content across different plastid regions.
Phylogenomic analyses
We generated 22 data matrices (Table 1) that differed in the types of plastid DNA data they contained, the approach used to find and remove potentially saturated loci, and the degree of gap removal. The alignment lengths, numbers of parsimony informative sites, AT contents, and best-fitting substitution models of all 22 matrices are shown in Table 1. We used the alignments to infer 22 maximum likelihood (ML) trees. The ML trees that resulted from an unpartitioned strategy were largely congruent with those inferred using a partitioned strategy (Figs. 1, and S2, S3, S4, S5, and S6). In the saturation analysis, IQ-TREE v1.6.12 [27] failed to build gene trees for 45 out of the 242 loci because of their lack of parsimony-informative sites (Table S2). Thus, only 197 loci and their gene trees were used to calculate patristic distance (PD) and p in TreSpEx v1.1 [28]. In the TreSpEx analysis, 55 loci exhibited possible saturation based on slope (Fig. S7a), and a slightly different set of 50 loci showed possible saturation based on R2 values (Fig. S7b). Consequently, we performed two independent ML analyses with the 55 (CP-slope) and 50 genes (CP-R2) removed, but we found that the resulting topologies (Figs. S8 and S9) were largely congruent with that based on complete plastomes (CP). Moreover, increasing the percentage of gaps in alignments did not affect tree topology (data not shown, but previously made available at Dryad [29]). The number of gaps among the alignments of 242 loci were highly variable, ranging from zero (e.g., atpB) to 28,413 (ccsA-trnLUAG) (Fig. S10).

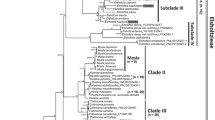
Maximum likelihood tree of tribe Perseeae based on unpartitioned complete plastomes (CP). The support values of Shimodaira-Hasegawa-like approximate likelihood ratio test (SH-aLRT ≥ 80%; on the left) and ultrafast bootstrap (UFBS ≥ 95%; on the right) are shown on branches. “-” above branches indicate SH-aLRT < 80% and UFBS < 95%, respectively
All 22 ML tree topologies were largely congruent; we therefore present only the unpartitioned CP ML tree in the main text (Fig. 1), and we regard this tree as the species tree. All trees revealed that tribe Perseeae is a monophyletic group comprised of six strongly supported major clades based on Shimodaira-Hasegawa-like approximate likelihood ratio tests (SH-aLRTs) and ultrafast bootstrap (UFBS) (i.e., SH-aLRT = 95.3–100%, UFBS = 97–100%; Fig. 1). In clade I, Alseodaphne gracilis Kosterm., A. huanglianshanensis H.W. Li & Y.M. Shui, and A. semecarpifolia Nees formed a monophyletic group that was sister to Dehaasia incrassata Kosterm. and Nothaphoebe umbelliflora Bl., and Persea borbonia Spreng., a species distributed in the southeastern United States, was sister to the rest of the clade. Clade II contained Persea americana Mill., which is native to Central America, and its unique phylogenetic position indicates that Persea Mill. is not monophyletic. Clade III comprised three species of Alseodaphnopsis H.W. Li & J. Li: A. andersonii H.W. Li & J. Li, A. petiolaris H.W. Li & J. Li, and A. sichourensis H.W. Li & J. Li. In clade IV, Nothaphoebe cavaleriei H. Lév. was nested within the genus Phoebe. Clade V consisted of Alseodaphnopsis hainanensis H.W. Li & J. Li, A. rugosa H.W. Li & J. Li, and Alseodaphne yunnanensis Kosterm. Species of Machilus formed clade VI, which was sister to clade V.
Although the 22 ML trees were largely in agreement, the phylogenetic positions of several species of Machilus were unstable and showed low support (i.e., SH-aLRT < 80% or UFBS < 95%; Figs. 1, S2, S3, S4, S5, S6, S8 and S9).
Divergence time estimation
Based on the application of criteria for clock-likeness, reasonable gene tree lengths, and concordance with the species tree, the genes ycf1, ycf2 and ndhF were selected for molecular dating analyses. In an analysis in which we calibrated the stem node of clade IV (node 3, scenario 5; Table S3) using the fossil Machilus maomingensis Jin & Tang, we estimated the stem age of tribe Perseeae to be 57.69 million years old (Ma) (95% highest posterior density (HPD) = 46.96–68.39 Ma). Similarly, when we calibrated the stem node of clade III (node 4, scenario 3; Table S3) using the fossil Alseodaphne changchangensis Jin & Li, the stem age of tribe Perseeae was inferred to be 54.80 Ma (95% HPD = 46.0–64.57 Ma). These two independent dating analyses both yielded stem ages for tribe Perseeae close to 55.3 Ma, which we regard as an external standard based on a prior study (see Methods). Therefore, we used both M. maomingensis and A. changchangensis in a final dating analysis (nodes 3 and 4, scenario 6; Table S3).
Our final dating analysis in BEAST v2.6.3 [30] indicated that tribe Perseeae originated in the very late Paleocene (56.50 Ma; 95% HPD = 45.87–66.57 Ma; Fig. S11). The stem and crown age of Phoebe were estimated to be 35.82 Ma (95% HPD = 34.09–38.06 Ma) and 25.86 Ma (95% HPD = 16.73–34.77 Ma), respectively. The stem and crown age of Machilus were 26.33 Ma (95% HPD = 16.93–34.23 Ma) and 15.38 Ma (95% HPD = 8.65–23.9 Ma), respectively.
Diversification rate and shift
Speciation rates obtained from Bayesian analysis of macro-evolutionary mixtures (BAMM) v2.5 [31] indicated diversification rate heterogeneity within tribe Perseeae (Fig. 2a). One diversification rate shift was detected within clade VI (near node 1, ca. 11 Ma; Fig. 2b) by sampling the maximum posterior configuration with the highest frequency (f = 0.29; Figs. 2a and S12). The rate-through-time plot suggested that the global speciation and net diversification rates of tribe Perseeae accelerated at ca. 11 Ma, followed by a steady increase from 11 Ma to 5.5 Ma and a rapid increase at ca. 5.5 Ma (Fig. 3b). Speciation and net diversification rates of Machilus increased rapidly, particularly at 11 Ma and 5.5 Ma (Fig. 3c). After excluding Machilus, speciation and net diversification rates of tribe Perseeae increased steadily and slowly from 46.48 Ma to the present (Fig. 3d). The diversification rate patterns and locations of rate shifts of tribe Perseeae were consistent under different global sampling probabilities (Figs. 2a and S13).

Combined diversification rate analyses and ancestral habitat reconstruction of tribe Perseeae. (a) Phylorate inferred from BAMM analysis with “ExpectedNumberofShifts” set to 1 and global sampling probability set to 0.135. (b) Ancestral habitat reconstructed by R package phytools with SYM model. Speciation rates (lineages per million years) are color-coded on branches in (a). Red circles in (a) indicates the position of speciation rate shift. Numbers in the circles near the nodes in (b) correspond to those node numbers listed in the main text and Fig. S11. The pie charts at the nodes represent the posterior probabilities of ancestral habitat types. Dated phylogeny of tribe Perseeae is derived from Fig. S11. Pl and Qu are abbreviations for Pliocene and Quaternary, respectively

Speciation, extinction, and net diversification rates through time plots obtained from the BAMM analyses. (a) The global temperature change in the past 45 Ma is derived from Zachos et al. [113]. Plots (b), (c) and (d) are diversification rates for Perseeae, Machilus, and Perseeae without Machilus, respectively; speciation, extinction, and net diversification rates are colored in red, black, and green, respectively; the shaded areas denote 95% confidence intervals in rate reconstructions
The speciation and extinction rates obtained using CPP on Mass-Extinction Times (CoMET) showed a pattern highly similar to that obtained using BAMM (Fig. S14). Specifically, the speciation rate accelerated at ca. 11 Ma and experienced a significant increase at 5.5 Ma (Fig. S14a). Moreover, we detected no mass extinction events. Within the CoMET analysis, the effective sample sizes (ESSs) of all parameters were greater than 200, and the Geweke statistic plots tended towards satisfactory, indicating reliability (Fig. S15).
Ancestral habitat
Among the 52 sampled species of tribe Perseeae, 17 inhabit only tropical forests, 17 are endemic in the East Asian subtropical EBLFs, 17 are found in both tropical forests and the East Asian subtropical EBLFs, and one occurs in the southeastern evergreen forests of North America (Table S4). Thus, we coded the species according to their modern-day habitats within these three major vegetation types and reconstructed the ancestral habitat using stochastic character mapping (SIMMAP) [9]. In SIMMAP, we used the “symmetric” model (SYM), which was the best-fitting model according to the Akaike Information Criterion (AIC) (Table S5). Our analyses revealed that the most recent common ancestors (MRCAs) of Phoebe and Machilus occurred in tropical forests in the Oligocene and Miocene, respectively (Fig. 2b). Phoebe and Machilus began colonizing the East Asian subtropical EBLFs at ca. 21 Ma and 11 Ma, respectively (Fig. 2b).
Discussion
Phylogenetic relationships within tribe Perseeae
Previous studies have suggested that conflicting phylogenetic signals among different plastid regions and different strategies for data partitioning, and substitution saturation are all factors that can affect tree topologies [32,33,34]. Here, however, we found that the phylogenetic relationships of the six clades (clades I–VI) and the species relationships within clades I–V were robust to these factors across 22 ML tree reconstructions. This result strongly suggests that these factors had no or negligible influence on the tree topology of tribe Perseeae.
Gaps in sequences are thought to have especially strong effects on tree topologies and phylogenetic accuracy in some cases, although the exact nature of the relationship between gaps and phylogenetic inference remains somewhat unclear. For example, Duvall et al. [35] noted that alignment gaps often occur at sites of ambiguous poly-A/T sequences. Thus, these sequences, in particular, may affect phylogenetic inference. Consistent with Duvall et al. [35], we found that AT content decreased (from 61.7 to 61.4%) as gap content was reduced (from 90% gaps to 0% gaps) in our sequence matrices (Table 1). Aside from specific types of gaps, the distribution and complexity of gaps may also affect phylogenetic accuracy [36], and our data (CP) exhibited complex gap distributions (Fig. S10). However, topologies of 14 datasets representing different levels of gappy-ness (from CP-gappy90 to CP-gappy0; Table 1) all agreed with the topology of the CP ML tree, suggesting that neither ambiguous poly-A/T sequences nor the complex distribution of gaps influenced our tree topology.
Phylogenetic relationships of the six clades revealed by the CP tree (and other trees) were consistent with the coalescent-based plastome tree generated in ASTRAL III [37] by Liu et al. [26]. However, the ASTRAL plastome tree from that study and our CP tree were highly incongruent with the nuclear ribosomal DNA (nrDNA) ML tree of Liu et al. [26], which showed that clades III and V formed a strongly supported group (BS = 95%) that was sister to clade IV (BS = 79%). This differs from our result, in which clade VI is sister to clade V and clade III is sister to clades IV + V + VI. Significant incongruence between plastome and nrDNA phylogenies has been observed for many plant groups, such as Heuchera L. (Saxifragaceae) [38], the Amelanchier-Malacomeles-Peraphyllum clade (Rosaceae) [39], and Apioideae (Apiaceae) [40, 41]. Such incongruence may be attributed to sampling error, paralogy of nrDNA sequences (due to allopolyploidy or incomplete concerted evolution), incomplete lineage sorting (ILS), or hybridization and introgression [42, 43]. Similar to the nrDNA-ML tree of Liu et al. [26], previous phylogenetic studies using different species, accessions, and molecular markers (ITS + LEAFY intron II) revealed that clade V was closer to clade III rather than clade VI [19, 21]. Therefore, sampling error and paralogy of nrDNA may not cause incongruence in tribe Perseeae, and, notably, polyploids have not been found in the tribe to date [44]. Although ILS and hybridization/introgression can now be modeled using coalescent and network approaches, respectively, disentangling the two evolutionary phenomena remains challenging [45].
Nothaphoebe cavaleriei nested within Phoebe in our study, consistent with the results of Rohwer et al. [20]. Thus, we agree with the taxonomic treatment of N. cavaleriei within Phoebe [46]. In our study, Phoebe was divided into three subclades (subclades A, B, and C) with high support, but there were no obvious morphological synapomorphies uniting each clade. Morphological characters, such as the absence, presence, or type of hairs on outside tepals, have been traditionally used to delimit intrageneric sections within Phoebe [18]. However, the three subclades identified here are inconsistent with these morphologically defined sections. For example, P. chekiangensis C.B. Shang from sect. Caniflorae Meissn. was not grouped with P. glaucophylla H.W. Li and P. macrocarpa C.Y. Wu from the same section (Fig. 1).
In the genus Machilus, our phylogenetic results showed three strongly supported subclades (subclades D, E, and F). However, resolution within subclade F was poor, and this may be because of recent differentiation or decreased substitution rate in this subclade [20]. Thus, resolving the relationships within subclade F may require a large set of nuclear markers and approaches that utilize and visualize even weak signals in the data, such as simplex plots of tree concordance [47] and the D-statistic (i.e., “ABBA BABA”) [48].
The robustness of diversification analysis with BAMM
Although incomplete sampling can potentially affect diversification rate analyses [49], BAMM has been shown to perform well under complete to low taxon sampling [50, 51]. To test the sensitivity of BAMM results to incorrect estimates of species diversities and incomplete sampling, Shi and Rabosky [52] ran an additional analysis with sampling probability halved and found that diversification rates were extremely similar under different sampling probabilities. We implemented a similar strategy and found that global sampling probability did not affect our results. Therefore, we have confidence in the results generated by BAMM. Nevertheless, our sampling of tribe Perseeae remains limited and it should be noted that biased taxon samplings (e.g., phylogenetic bias and spatial bias) can potentially lead to biasness in diversification rate patterns, which are inherent in large-scale biodiversity analyses [53,54,55,56].
The expansion of subtropical EBLFs in the early Miocene
Communities and biomes respond to climate change through species dynamics [57]. Thus, the dynamics of Lauraceae, such as tribe Perseeae, may provide insights into the overall development of subtropical EBLFs because they are keystone species. Here, we found that tribe Perseeae did not colonize the subtropical EBLFs until the O-M boundary (21.77 Ma; Fig. 2), indicating that modern subtropical EBLFs of East Asia may not have originated until this time. Our result is congruent with the conclusion of Yu et al. [13] based on inferences from Theaceae, which also contains many keystone EBLF species. Other essential elements of the East Asian subtropical EBLFs, such as Magnolia L. sect. Yulania Dandy [58], also appear to have originated around the O-M boundary.
Lauraceae species were geographically widespread throughout the Late Cretaceous to early Eocene based on their common occurrence in fossil assemblages from North America, Europe, and Asia [59,60,61]. Fossils of tribe Perseeae and its sister groups (tribe Laureae and tribe Cinnamomeae) in the Eocene indicate that these tribes once colonized high latitudes of the Northern Hemisphere (e.g., the London Clay Flora [62], west and east Beringia [63]). Beringia was occupied by deciduous and evergreen broad-leaved forests (the boreotropical flora) in the Eocene [64], suggesting that tribe Perseeae colonized temperate or subtropical forests. As a response to dramatic climate change from the middle Eocene to the Oligocene, the boreotropical forests migrated to low latitude regions, and many temperate/subtropical lineages became extinct.
There was a broad arid/semi-arid belt between 20–40°N in East Asia from at least the Eocene [65]. The O-M boundary is broadly characterized by a brief glaciation in the Southern Hemisphere and global cooling [66], which, in eastern Asia, led to changes in moisture patterns [67] and in floral composition since the early Miocene [68]. The increase in humidity supported new, moist forests [69] and, thus, the immigration of plants from the lower-latitude paleotropics to new, higher-latitude, subtropical habitats [70]. It was not until the Neogene that the Lauraceae became common and dominant in the East Asian fossil flora [71, 72]. This is consistent with our ancestral habitat reconstruction results, which suggest that the MRCA of Machilus and Phoebe occurred in tropical forests (Fig. 2b) and that an ancestor of Phoebe colonized the East Asian subtropical EBLFs beginning at ca. 21 Ma, around the O-M boundary. The migration of Machilus into the EBLFs came later, around 11 Ma, and may be more consistent with a peak intensification of the EAM [73]. Overall, our results, if they can be applied broadly to the EBLFs, suggest that this biome underwent at least two major periods of diversification, first at the O-M boundary and again during a period of particularly strong regional monsoons.
Rapid species accumulation of East Asian subtropical EBLFs in the late Miocene
Results from BAMM supported two periods of increased diversification rate within tribe Perseeae in the late Miocene (ca. 11 Ma and 5.5 Ma; Figs. 2a and 3). The two accelerations of diversification rate in the late Miocene were consistent with the CoMET analysis (Fig. S15). Thus, our results indicated that the late Miocene was pivotal for the assembly and evolution of tribe Perseeae and, by proxy, possibly for the modern subtropical EBLFs [14]. Notably, the acceleration in diversification ca. 5.5 Ma may coincide with a slight decrease in the intensity of the EAM [73], which may have opened up additional niches within the developing ELBFs.
High species diversity, such as that found in the EBLFs, may result from recent rapid speciation in a ‘cradle’ and/or the gradual accumulation and preservation of species over time in a ‘museum’. Some species within the EBLFs may be relicts from the early Cretaceous or late Jurassic period that survived in glacial refugia within hilly or mountainous regions of southern or eastern China where the EBLFs now occur [74]. However, unlike species of tribe Perseeae, most of these relicts are monotypic in subtropical EBLFs. By contrast, rapid species accumulation in the late Miocene has been reported for several dominant groups in subtropical EBLFs, such as Schima [13], Camellia L. (Theaceae) [13], and Quercus sect. Cyclobalanopsis [8].
Increased annual precipitation has sometimes been invoked to explain the higher diversification rates of plants and the rapid assembly of the East Asian subtropical EBLFs in the late Miocene (e.g., [13, 14, 75]). For example, Farnsworth et al. [76] indicated that a “super monsoon” existed from ca. 12 to ca. 4 Ma, resulting in increased annual precipitation. This is consistent with studies based on magnetic records and isotopic evidence that also support a progressively intensified EAM in the late Miocene and early Pliocene [77, 78]. Although the correlation between the intensified EAM and greater precipitation and increased diversification rates remains unclear, annual rainfall is the single best water-related predictor for species richness of Chinese woody plants [79]. Thus, the intensified rainfall associated with the EAM may have promoted periods of rapid assembly of the East Asian subtropical EBLFs in the late Miocene [13, 14, 75], consistent with our results for tribe Perseeae. However, fluctuations in the intensity of the monsoon may have opened additional niches through time and supported additional diversification.
Conclusions
Our phylogenetic analyses have revealed six strongly-supported major clades within tribe Perseeae (Lauraceae) based on plastomes. Machilus and Phoebe, two dominant genera in the East Asian subtropical EBLFs, were monophyletic. Our results indicate that tribe Perseeae from tropical forests in the late Paleogene colonized the East Asian subtropical EBLFs in the early Miocene, and this was possibly facilitated by changes in global moisture patterns, including periods of change to the intensity of the EAM. In general, increases in moisture and humidity probably supported diversification in lineages of tribe Perseeae within the EBLFs. Along with prior results from Theaceae [13], which is also a dominant element of the ELBFs, our results suggest that the Miocene was a critical period for the assembly of the modern ELBFs. Moreover, our study illustrates how tribe Perseeae evolved under past climatic changes and, therefore, may provide insights into the possible responses of subtropical EBLFs to ongoing and future climate change.
Methods
Sampling, DNA sequencing, plastome assembly and annotation
We used a total of 43 samples for DNA sequencing in this study. These samples represented 37 species from four genera in tribe Perseeae and two species from tribe Cinnamomeae as an outgroup (Table S6). We used silica-gel-dried leaf tissues for DNA extraction. The first author identified and deposited voucher specimens in the herbarium of the South China Botanical Garden, Chinese Academy of Sciences (IBSC) (Table S6). We collected the plant materials from the wild and from botanical gardens. No specific permissions or licenses were required for our collections and experiments.
For DNA extraction, sequencing, plastome assembly, and annotation, we followed the methods of Xiao et al. [32]: we extracted genomic DNA using the cetyl trimethyl ammonium bromide (CTAB) method [80] and assembled and annotated plastomes with NOVOPlasty v2.7.2 [81] and GeSeq [82], respectively. We generated maps of annotated plastomes with the OrganellarGenomeDRAW tool (OGDRAW; [83]).
Data preparation and phylogenetic analyses
In addition to newly sequenced plastomes, we also downloaded 17 plastomes representing 15 species of tribe Perseeae and one species of tribe Cinnamomeae (Table S7) from the National Center for Biotechnology Information (NCBI) [84] for use in phylogenetic analyses. Thus, our samples for phylogenetic reconstruction comprised 58 plastomes of tribe Perseeae, representing 53 species, and three plastomes of outgroup taxa, for a total of 61 plastid genomes. From all plastomes (newly sequenced and downloaded), we extracted protein-coding genes (PCGs), introns, intergenic spacers (IGSs), tRNAs, and rRNAs using a python script [85] and aligned the extracted loci separately using MAFFT v7.467 [86] in “localpair” mode with 1000 iterative refinements.
Inaccurate tree inference can arise for both statistical and biological reasons [87], including conflicting phylogenetic signals between coding and non-coding regions [32]; differences in data partitioning [33]; nucleotide substitution saturation [34], and gap percentages [35, 36]; and other causes. Therefore, we generated sequence matrices and performed phylogenetic analyses to evaluate the potential effects of these factors, as described below.
We generated three sequence matrices, PCG, NPCG (non-protein coding genes), and CP (complete plastomes with one inverted repeat region removed to avoid redundancy), to investigate potentially conflicting phylogenetic signals between coding and non-coding regions of the plastomes [32]. Initially, we concatenated loci to construct each matrix using the python script AMAS v1.0 [88], and we then manually edited the matrices with BioEdit v7.2.5 [89] when necessary.
Partitioning has been shown to improve phylogenetic inference in some studies [33] and has had no impact in others [32, 90]. We therefore implemented both partitioned and unpartitioned strategies on the three sequence matrices (CP, PCG, and NPCG) to determine the effects of partitioning. After generating the partition files in AMAS, we manually modified them into RAxML style, treating each locus as an independent data block. We used “-spp” in IQ-TREE v1.6.12 to determine the partition scheme [27, 91].
To evaluate the potential effect of nucleotide substitution saturation, we identified loci with high saturation levels using TreSpEx v1.1 [28]. The rationale of TreSpEx is that the larger the slope or R2 in a linear regression analysis of PD and uncorrected genetic distance (p), the less saturated the locus. Therefore, we first calculated PD and p based on the gene trees before performing linear regression in TreSpEx. We then plotted the distribution of slope and R2 values in R v3.6.2 [92]. We considered loci located on the shoulders of the slope or R2 plots as possibly saturated. Thus, we generated two new sequence matrices, CP-slope and CP-R2, with the potentially saturated loci removed.
As Duvall et al. [35] indicated, gaps in alignments can affect tree topology. Therefore, we trimmed sites above certain thresholds of gap percentage from the CP matrix using ClipKIT [93]. The trimming mode was set to “gappy”, and the threshold was set to 0.90 (removing sites with > 90% gap percentage), 0.70, 0.50, 0.25, 0.15, 0.14, 0.12, 0.10, 0.09, 0.07, 0.05, 0.04, 0.02, or 0 (including no gaps). The 14 gap-stripped matrices were used for ML analyses.
Combined, all of our strategies yielded 22 data matrices for analysis (Table 1). These included six matrices containing different plastid regions, with or without partitioning (CP, PCG, NPCG, CP-p, PCG-p, and NPCG-p). Among the 22 matrices, two (CP-slope and CP-R2) were generated by removing probably saturated loci. The remaining 14 matrices were generated by trimming gapped sites of CP that were above specific thresholds of gap percentage (“CP-gappy” in Table 1).
For all 22 matrices, we determined alignment length, number of gaps, parsimony-informative sites, and AT content using AMAS. We plotted the variation of gap abundance across loci in R. For each matrix or partition, we determined the best-fitting models of nucleotide substitution in ModelFinder [94] under the Bayesian Information Criterion (BIC) implemented in IQ-TREE. We used IQ-TREE to perform ML analyses with 1000 replicates each of UFBS [95] and the SH-aLRT [96] to determine node support. In all analyses, we applied the ‘-bnni’ parameter to reduce the risk of overestimating branch support due to any possible severe model violations [27].
Divergence time estimation
For divergence time estimation, we downloaded an additional 17 plastomes representing other major clades of Lauraceae (Table S7). The large volume of plastid data could make divergence-time estimation intractable, and among loci, topology and substitution rate heterogeneity may lead to model violations. Thus, we applied the gene-shopping method to filter protein-coding genes using SortaDate [97] based on three principles: clock-likeness, reasonable tree length, and least topological conflict with the species tree (the unpartitioned CP ML tree; Fig. 1).
We performed molecular dating analysis in BEAST v2.6.3 [30]. For the analysis, we concatenated the selected loci, used BEAUti [30] to import them, set the substitution model to GTR, implemented a relaxed log-normal molecular clock, and applied a Yule model for the tree branching process with birth rate set to a gamma distribution with an alpha and beta of 0.001 and 1000, respectively. We ran the analysis for 200,000,000 Markov chain Monte Carlo (MCMC) generations, sampling every 20,000 generations. Following the analysis, we imported the log file into Tracer v1.7.1 [98] to verify stationarity based on all estimated parameters with ESS ≥ 200. After removing the first 20% of trees as burn-in, we generated a maximum clade credibility (MCC) tree in TreeAnnotator v2.6.3 [30] and visualized it using FigTree v1.4.3 [99].
We used a secondary calibration point and three macrofossils for divergence time estimation. For the secondary calibration, we set a normal prior with an offset of 98 Ma, a mean of 0, and a sigma of 1.0 for the crown age of Lauraceae following Nie et al. [100] and Li et al. [101]. One of the macrofossils, Neusenia tetrasporangiata Eklund, is a well-preserved flower bud from the Santonian/Campanian (ca. 83 Ma; late Cretaceous) in the Neuse River locality in North Carolina, USA, and it can be assigned to Neocinnamomum H. Liu [102, 103]. Therefore, we used this fossil to set a log-normal prior with an offset of 83 Ma, a mean of 1.0, and a standard deviation of 0.6 for the crown age of Neocinnamomum-Caryodaphnopsis-core Lauraceae following the strategy of Li et al. [104].
Two leaf macrofossils are also highly relevant to divergence time dating in tribe Perseeae: Alseodaphne changchangensis Jin & Li, which was found in the Upper Member, or rock bed assemblage, of the Eocene Changchang Formation of the Changchang Basin within Hainan Province, China [105], and Machilus maomingensis Jin & Tang, which was found in the Upper Member of the middle/late Eocene Youganwo Formation outcropping in the Maoming Basin of Guangdong Province, China [106]. Determining the genera of fossils from Lauraceae and extant groups is difficult based only on isolated leaves [107], unless the species is clearly recognizable [44]. Thus, based on the CP ML tree (Fig. 1), there were several options for the placement of A. changchangensis and M. maomingensis for age calibration. We considered three nodes (nodes 2, 3 and 4 in Fig. 2b and S11) as candidates for calibration points. In scenarios 1, 2, and 3, we placed A. changchangensis at nodes 2, 3, and 4, respectively (Table S3). In scenarios 4 and 5, we fixed M. maomingensis at nodes 2 and 3, respectively (Table S3).
Fossil fruit cupules attributable to the Perseeae + Laureae clade are abundant in the London Clay Flora, which represents the early Eocene beginning 56 Ma [62]. Thus, the stratigraphic age of London Clay Flora has been considered to be the minimum stem age of tribe Perseeae, which was estimated to be 55.3 Ma (95% HPD = 41.4–69.9 Ma) by Li et al. [21] using molecular dating. Therefore, we ran the dating scenarios with the different calibration points for A. changchangensis and M. maomingensis and determined their placement based on the result showing a stem age for the tribe closest to 55.3 Ma. We then used the inferred best calibrations for both fossils in a final dating scenario, Scenario 6 (Table S3).
In scenarios 1–6, we fixed the position of N. tetrasporangiata and the secondary calibration point at nodes 5 and 6, respectively (Table S3; Fig. S11). For A. changchangensis, we applied a log-normal prior distribution with an offset of 37 Ma, a mean of 1.0 and, a standard deviation of 0.85, and for M. maomingensis, we used a log-normal prior distribution with an offset of 33.7 Ma, a mean of 1, and a standard deviation of 0.85. In the molecular dating analyses, we found that the phylogenetic positions of several species of Phoebe had changed, although they were strongly supported in the ML tree (Fig. 1). This may have been caused by reduced numbers of parsimony informative sites in the three selected loci. To reduce their influence on tree topology and downstream analyses, we constrained the phylogenetic positions of these Phoebe species in scenarios 1–6.
Diversification rate analysis
Although no method of diversification rate analysis is entirely robust to poor taxon sampling, BAMM v2.5 [31] has been shown to be less sensitive than RPANDA [108] and the DR statistic [109] with a moderate level of missing taxa [53]. We, therefore, employed BAMM to infer speciation rates and possible rate shifts across the dated MCC tree. Before applying BAMM, we removed outgroup taxa using APE v5.4-1 [110] and determined priors with BAMMtools v2.1.6 [111] in R. The prior for “ExpectedNumberofShifts” was set to 1 because the tip number of the MCC tree was smaller than 500 [111]. To account for missing extant taxa, we specified the global sampling probability as 0.135. We ran BAMM for 10,000,000 generations and sampled every 1000 generations. Following the analysis, we calculated the ESS of loglikelihood and number of shifts to evaluate convergence based on ESS ≥ 200 using the CODA R package [112]. Thereafter, we analyzed BAMM output files using BAMMtools with 25% of samples discarded as burn-in. From the remaining samples, we inferred the 95% credible set of macroevolutionary rate shift configurations according to BAMM posterior probability, and we used this to plot speciation rate, extinction rate, and net diversification rate through time in BAMMtools. Within the plots, we included relative global temperature change during the past 45 million years (Fig. 3a) according to Zachos et al. [113].
Although we have collected 365 accepted species names of tribe Perseeae (Table S1), the true diversity of this tribe may be larger, because of inadequate investigation and taxonomic difficulties. To test the sensitivity of BAMM analysis to inaccurate estimates of diversity in tribe Perseeae and incomplete sampling, we ran an additional analysis using BAMM with global sampling probability halved and other prior parameters unchanged, following Shi and Rabosky [52].
To evaluate the robustness of the diversification pattern inferred by BAMM, we also performed an analysis using CoMET within the TESS R package [114]. Before running CoMET, we specified the prior distribution of model parameters following Höhna et al. [115], and we used the same sampling probability and input tree as in the BAMM analysis. We ran the CoMET analysis for 4,000,000 iterations, with 400,000 iterations discarded as burn-in. We summarized and visualized the results using the tess.process.output and tess.plot.output functions, respectively. To ensure reliable estimates, we performed single-chain diagnostics to evaluate the ESS and Geweke diagnostic for diversification rates, shift times, and mass extinction events.
Ancestral habitat
We determined the geographic distribution of all sampled species of tribe Perseeae based on the National Specimen Information Infrastructure (NSII) [116] and the Flora of China [18] and used the distributions to assess their vegetation types [15, 117]. Based on the results, we identified three vegetation types and coded species according to their presence in each: (1) tropical forests, (2) East Asian subtropical EBLFs, and (3) evergreen forests of the southeastern United States of America (U.S.A.). Tropical forests in Asia and the Americas were included in the same category. We coded species found in both tropical forests and subtropical EBLFs as an additional unique state.
As incomplete sampling of outgroups can bias ASR, we excluded all outgroups before analysis [13, 118]. We reconstructed the ancestral habitat of tribe Perseeae using SIMMAP [9], a Bayesian approach implemented in the R package phytools [119]. For the analysis, we selected the best-fitting model from among “equal-rates” (ER), “symmetric” (SYM), and “all-rates-different” (ARD) according to the AIC using the function fitDiscrete in the R package geiger [120]. Within SIMMAP, we set the number of simulations to 200, and we visualized the analysis results in the ggtree R package [121].
Availability of data and materials
Plastomes generated in this study are released in the Science Data Bank (https://www.scidb.cn/) [122] and NCBI (https://www.ncbi.nlm.nih.gov/nuccore/) [84], with GenBank accession numbers shown in Table S6. Sequencing data are deposited in Sequence Read Archive of NCBI (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA756055), the SRA accession numbers are SRR15566192-SRR15566234 (Table S6). All the sequence matrices in this study, partition files, and generated tree files are deposited at Dryad repository [29]. Public access to the databases mentioned above are open and no administrative permissions are needed for accessing and using the data. Material samples are available from authors. Voucher specimens were deposited in the herbarium of the South China Botanical Garden (IBSC) (Table S6).
Abbreviations
- AIC:
-
Akaike Information Criterion
- ASR :
-
Ancestral state reconstruction
- BAMM :
-
Bayesian analysis of macro-evolutionary mixtures
- BIC :
-
Bayesian Information Criterion
- CP :
-
Complete plastomes
- CoMET :
-
CPP on Mass-Extinction Times
- CTAB :
-
cetyl trimethyl ammonium bromide
- EAM :
-
East Asian monsoon
- EBLFs :
-
Evergreen broad-leaved forests
- ESS :
-
Effective sample size
- HPD :
-
Highest posterior density
- IBSC :
-
The herbarium of South China Botanical Garden, Chinese Academy of Sciences
- IGS :
-
Intergenic spacers
- ILS :
-
Incomplete lineage sorting
- IR:
-
Inverted repeat region
- LSC :
-
Large single-copy region
- MCC :
-
Maximum clade credibility
- ML :
-
Maximum likelihood
- MRCA :
-
Most recent common ancestor
- NCBI:
-
National Center for Biotechnology Information
- NPCG:
-
Non-protein coding genes
- nrDNA:
-
Nuclear ribosomal DNA
- NSII:
-
National Specimen Information Infrastructure
- O-M:
-
Oligocene-Miocene
- OGDRAW:
-
OrganellarGenomeDRAW
- PCG:
-
Protein coding genes
- PD:
-
Patristic distance
- rRNA:
-
Ribosomal RNA
- SH-aLRT:
-
Shimodaira-Hasegawa-like approximate likelihood ratio test
- SSC:
-
Small single-copy region
- tRNA:
-
Transfer RNA
- UFBS:
-
Ultrafast bootstrap
References
Slik JWF, Franklin J, Arroyo-Rodríguez V, Field R, Aguilar S, Aguirre N, et al. Phylogenetic classification of the world’s tropical forests. Proc Natl Acad Sci USA. 2018;115(8):1837–1842.
Yang Y, Fujihara M, Li B, Yuan X, Hara K, Da L, et al. Structure and diversity of remnant natural evergreen broad-leaved forests at three sites affected by urbanization in Chongqing metropolis, Southwest China. Landsc Ecol Eng. 2014;10(1):137–149.
Song Y-C. On the global position of the evergreen broad-leaved forests of China. In: Box EO, Peet RK, Masuzawa T, Yamada T, Fujiwara K, Maycock PF, editors. Vegetation science in forestry. Netherlands: Springer; 1995. p. 69–84.
Zhang J, Chu Z, Ge Y, Zhou X, Jiang H, Chang J, et al. TRIPLEX model testing and application for predicting forest growth and biomass production in the subtropical forest zone of China’s Zhejiang Province. Ecol Model. 2008;219(3):264–275.
Mucina L. Biome: evolution of a crucial ecological and biogeographical concept. New Phytol. 2019;222(1):97–114.
Losos JB. Phylogenetic perspectives on community ecology. Ecology. 1996;77(5):1344–1354.
Zhao J, Li Q. Global environmental changes and their impact on biological evolution during the Oligocene-Miocene transition. SCI SIN Vitae. 2019;49:902–915.
Deng M, Jiang XL, Hipp AL, Manos PS, Hahn M. Phylogeny and biogeography of East Asian evergreen oaks (Quercus section Cyclobalanopsis; Fagaceae): Insights into the Cenozoic history of evergreen broad-leaved forests in subtropical Asia. Mol Phylogenet Evol. 2018;119:170–181.
Bollback JP. SIMMAP: Stochastic character mapping of discrete traits on phylogenies. BMC Bioinform. 2006;7(1):88.
Pagel M. Detecting correlated evolution on phylogenies: a general method for the comparative analysis of discrete characters. Proc Royal Soc B. 1994;255(1342):37–45.
Höhna S, Landis MJ, Heath TA, Boussau B, Lartillot N, Moore BR, et al. RevBayes: Bayesian phylogenetic inference using graphical models and an interactive model-specification language. Syst Biol. 2016;65(4):726–736.
De-Nova JA, Medina R, Montero JC, Weeks A, Rosell JA, Olson ME, et al. Insights into the historical construction of species-rich Mesoamerican seasonally dry tropical forests: the diversification of Bursera (Burseraceae, Sapindales). New Phytol. 2012;193(1):276–287.
Yu XQ, Gao LM, Soltis DE, Soltis PS, Yang JB, Fang L, et al. Insights into the historical assembly of East Asian subtropical evergreen broadleaved forests revealed by the temporal history of the tea family. New Phytol. 2017;215(3):1235–1248.
Wang W, Xiang X-G, Xiang K-L, Ortiz RdC, Jabbour F, Chen Z-D. A dated phylogeny of Lardizabalaceae reveals an unusual long-distance dispersal across the Pacific Ocean and the rapid rise of East Asian subtropical evergreen broadleaved forests in the late Miocene. Cladistics. 2020;36(5):447–457.
Song Y-C, Da L-J. Evergreen Broad-Leaved Forest of East Asia. In: Box EO, editors. Vegetation structure and function at multiple spatial, temporal and conceptual scales. Switzerland: Springer International; 2016. p. 101–128.
Xiang K-L, Erst AS, Xiang X-G, Jabbour F, Wang W. Biogeography of Coptis Salisb. (Ranunculales, Ranunculaceae, Coptidoideae), an Eastern Asian and North American genus. BMC Evol Biol. 2018;18(1):74.
Yang T, Lu L-M, Wang W, Li J-H, Manchester SR, Wen J, et al. Boreotropical range expansion and long-distance dispersal explain two amphi-Pacific tropical disjunctions in Sabiaceae. Mol Phylogenet Evol. 2018;124:181–191.
Li H-W, Li J, Huang P-H, Wei F-N, Tsui H-P, van der Werff H. Lauraceae. In: Wu Z-Y, Raven PH, Hong D-Y, editors. Flora of China. Beijing & Saint Louis: Science Press & Missouri Botanical Garden; 2008.
Mo YQ, Li L, Li JW, Rohwer JG, Li HW, Li J. Alseodaphnopsis: A new genus of Lauraceae based on molecular and morphological evidence. PLoS One. 2017;12(10):e0186545.
Rohwer JG, Li J, Rudolph B, Schmidt SA, van der Werff H, Li H-w. Is Persea (Lauraceae) monophyletic? Evidence from nuclear ribosomal ITS sequences. Taxon. 2009;58(4):1153–1167.
Li L, Li J, Rohwer JG, van der Werff H, Wang Z-H, Li H-W. Molecular phylogenetic analysis of the Persea group (Lauraceae) and its biogeographic implications on the evolution of tropical and subtropical Amphi-Pacific disjunctions. Am J Bot. 2011;98(9):1520–1536.
National Specimen Information Infrastructure. https://www.nsii.org.cn. Accessed 15 March 2021.
Rohwer JG. Toward a phylogenetic classification of the Lauraceae: Evidence from matK sequences. Syst Bot. 2000;25(1):60–71.
Chanderbali AS, van der Werff H, Renner SS. Phylogeny and historical biogeography of Lauraceae: evidence from the chloroplast and nuclear genomes. Ann Mo Bot Gard. 2001;88(1):104–134.
Song Y, Yu WB, Tan YH, Jin JJ, Wang B, Yang JB, et al. Plastid phylogenomics improve phylogenetic resolution in the Lauraceae. J Syst Evol. 2020;58(4):423–439.
Liu Z-F, Ma H, Ci X-Q, Li L, Song Y, Liu B, et al. Can plastid genome sequencing be used for species identification in Lauraceae? Bot J Linn Soc. 2021;197(1):1–14.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2014;32(1):268–274.
Struck TH. TreSpEx–Detection of misleading signal in phylogenetic reconstructions based on tree information. Evol Bioinform. 2014;10:51–67.
Xiao T-W, Yan H-F, Ge X-J. Plastid phylogenomics of tribe Perseeae (Lauraceae) yields insights into the evolution of East Asian subtropical evergreen broad-leaved forests. Dryad. 2021. https://doi.org/10.5061/dryad.sqv9s4n4f.
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, et al. BEAST 2: a software platform for bayesian evolutionary analysis. PLoS Comp Biol. 2014;10(4):e1003537.
Rabosky DL. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS One. 2014;9(2):e89543.
Xiao T-W, Xu Y, Jin L, Liu T-J, Yan H-F, Ge X-J. Conflicting phylogenetic signals in plastomes of the tribe Laureae (Lauraceae). PeerJ. 2020;8:e10155.
Xi Z, Ruhfel BR, Schaefer H, Amorim AM, Sugumaran M, Wurdack KJ, et al. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc Natl Acad Sci USA. 2012;109(43):17519–17524.
Zhang R, Wang Y-H, Jin J-J, Stull GW, Bruneau A, Cardoso D, et al. Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Syst Biol. 2020;69(4):613–622.
Duvall MR, Burke SV, Clark DC. Plastome phylogenomics of Poaceae: alternate topologies depend on alignment gaps. Bot J Linn Soc. 2020;192(1):9–20.
Orton LM, Barberá P, Nissenbaum MP, Peterson PM, Quintanar A, Soreng RJ, et al. A 313 plastome phylogenomic analysis of Pooideae: Exploring relationships among the largest subfamily of grasses. Mol Phylogenet Evol. 2021;doi:https://doi.org/10.1016/j.ympev.2021.107110.107110.
Zhang C, Rabiee M, Sayyari E, Mirarab S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018;19(6):153.
Soltis DE, Kuzoff RK. Discordance between nuclear and chloroplast phylogenies in the Heuchera group (Saxifragaceae). Evolution. 1995;49(4):727–742.
Liu B-B, Campbell CS, Hong D-Y, Wen J. Phylogenetic relationships and chloroplast capture in the Amelanchier-Malacomeles-Peraphyllum clade (Maleae, Rosaceae): evidence from chloroplast genome and nuclear ribosomal DNA data using genome skimming. Mol Phylogenet Evol. 2020;147:106784.
Wen J, Xie DF, Price M, Ren T, Deng YQ, Gui LJ, et al. Backbone phylogeny and evolution of Apioideae (Apiaceae): New insights from phylogenomic analyses of plastome data. Mol Phylogenet Evol. 2021;161:107183.
Lee C-S, Downie SR. Phylogenetic relationships within Cicuta (Apiaceae tribe Oenantheae) inferred from nuclear rDNA ITS and cpDNA sequence data. Can J Bot-Rev Can Bot. 2006;84(3):453–468.
Acosta MC, Premoli AC. Evidence of chloroplast capture in South American Nothofagus (subgenus Nothofagus, Nothofagaceae). Mol Phylogenet Evol. 2010;54(1):235–242.
Rieseberg LH, Soltis D. Phylogenetic consequences of cytoplasmic gene flow in plants. Evol Trends Plants. 1991;5:65–84.
Rohwer JG. Lauraceae. In: Kubitzki K, Rohwer JG, Bittrich V, editors. Flowering Plants · Dicotyledons The Families and Genera of Vascular Plants, vol. 2. Berlin Heidelberg: Springer-Verlag; 1993. p. 366–390.
Morales-Briones DF, Liston A, Tank DC. Phylogenomic analyses reveal a deep history of hybridization and polyploidy in the Neotropical genus Lachemilla (Rosaceae). New Phytol. 2018;218(4):1668–1684.
Yang Y, Liu B. Species catalogue of Lauraceae in China: problems and perspectives. Biodiversity. 2015;23(02):232–236.
Allman ES, Mitchell JD, Rhodes JA. Gene tree discord, simplex plots, and statistical tests under the coalescent. Syst Biol. 2021;doi:https://doi.org/10.1093/sysbio/syab008.
Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Mol Biol Evol. 2011;28(8):2239–2252.
FitzJohn RG, Maddison WP, Otto SP. Estimating trait-dependent speciation and extinction rates from incompletely resolved phylogenies. Syst Biol. 2009;58(6):595–611.
Rabosky DL. BAMM at the court of false equivalency: A response to Meyer and Wiens. Evolution. 2018;72(10):2246–2256.
Meyer ALS, Wiens JJ. Estimating diversification rates for higher taxa: BAMM can give problematic estimates of rates and rate shifts. Evolution. 2018;72(1):39–53.
Shi JJ, Rabosky DL. Speciation dynamics during the global radiation of extant bats. Evolution. 2015;69(6):1528–1545.
Sun M, Folk RA, Gitzendanner MA, Soltis PS, Chen Z, Soltis DE, et al. Estimating rates and patterns of diversification with incomplete sampling: a case study in the rosids. Am J Bot. 2020;107(6):1–15.
Sun M, Folk RA, Gitzendanner MA, Soltis PS, Chen Z, Soltis DE, et al. Recent accelerated diversification in rosids occurred outside the tropics. Nat Commun. 2020;11(1):3333.
Zhang X, Landis JB, Sun Y, Zhang H, Lin N, Kuang T, et al. Macroevolutionary pattern of Saussurea (Asteraceae) provides insights into the drivers of radiating diversification. Proc Royal Soc B. 2021;288(1962):20211575.
Folk RA, Sun M, Soltis PS, Smith SA, Soltis DE, Guralnick RP. Challenges of comprehensive taxon sampling in comparative biology: Wrestling with rosids. Am J Bot. 2018;105(3):433–445.
McCarty JP. Ecological consequences of recent climate change. Conserv Biol. 2001;15(2):320–331.
Dong S-S, Wang Y-L, Xia N-H, Liu Y, Liu M, Lian L, et al. Plastid and nuclear phylogenomic incongruences and biogeographic implications of Magnolia s.l. (Magnoliaceae). J Syst Evol. 2021;doi:https://doi.org/10.1111/jse.12727.
Guo S-X, Li H-M. Late cretaceous flora from Hunchun of Jilin. Acta Palaeont Sin. 1979;18:547–559.
Friis EM, Crane PR, Pedersen KR. Early Flowers and Angiosperm Evolution. Cambridge: Cambridge University Press; 2011.
Takahashi M, Herendeen PS, Xiao X, Crane PR. Lauraceous fossil flowers from the Kamikitaba assemblage (Coniacian, Late Cretaceous) of Northeastern Japan (Lauraceae). Syst Bot. 2014;39(3):715–724.
Reid EM, Chandler MEJ. The London Clay Flora. London: British Museum (Natural History); 1933.
Graham A. The role of land bridges, ancient environments, and migrations in the assembly of the North American flora. J Syst Evol. 2018;56(5):405–429.
Tiffney BH. The Eocene North Atlantic Land Bridge: its importance in Tertiary and modern phytogeography of the Northern Hemisphere. J Arnold Arboretum. 1985;66(2):243–273.
Guo ZT, Sun B, Zhang ZS, Peng SZ, Xiao GQ, Ge JY, et al. A major reorganization of Asian climate by the early Miocene. Clim Past. 2008;4(3):153–174.
Zachos JC, Shackleton NJ, Revenaugh JS, Pälike H, Flower BP. Climate response to orbital forcing across the Oligocene-Miocene boundary. Science. 2001;292(5515):274.
Pfuhl HA, McCave IN: The Oligocene-Miocene boundary—cause and consequence from a Southern Ocean perspective. In: Deep-Time Perspectives on Climate Change: Marrying the Signal from Computer Models and Biological Proxies. Edited by Williams M, Haywood AM, Gregory FJ, Schmidt DN, vol. 2. London: Geological Society of London; 2007.
Chen Y-S, Deng T, Zhou Z, Sun H. Is the East Asian flora ancient or not? Natl Sci Rev. 2018;5(6):920–932.
Sun X, Wang P. How old is the Asian monsoon system?—Palaeobotanical records from China. Palaeogeogr, Palaeoclimatol, Palaeoecol. 2005;222(3):181–222.
Li X-C, Manchester SR, Xiao L, Wang Q, Hu Y, Sun B-N. Ormosia (Fabaceae: Faboideae) from the Miocene of southeastern China support historical expansion of the tropical genus in East Asia. Hist Biol. 2021;doi:https://doi.org/10.1080/08912963.2021.1877700.
Zhou Z-K, Huang J, Ding W-N. The impact of major geological events on Chinese flora. Biodivers Sci. 2017;25(2):123–135.
Editorial Committee of Cenozoic Flora of China. Plant Fossils of China, Vol. 3, Cenozoic Flora of China. Beijing: Science Press; 1978.
Ding W-N, Ree RH, Spicer RA, Xing Y-W. Ancient orogenic and monsoon-driven assembly of the world’s richest temperate alpine flora. Science. 2020;369(6503):578–581.
Axelrod DI, Al-Shehbaz IA, Raven PH. History of the modern flora of China. In: Zhang AL, Wu SG, editors. Floristic characteristics and diversity of East Asian plants. Beijing: China Higher Education; 1996. p. 43–55.
Chen X-H, Xiang K-L, Lian L, Peng H-W, Erst AS, Xiang X-G, et al. Biogeographic diversification of Mahonia (Berberidaceae): Implications for the origin and evolution of East Asian subtropical evergreen broadleaved forests. Mol Phylogenet Evol. 2020;151:106910.
Farnsworth A, Lunt DJ, Robinson SA, Valdes PJ, Roberts WHG, Clift PD, et al. Past East Asian monsoon evolution controlled by paleogeography, not CO2. Sci Adv. 2019;5(10):eaax1697.
Ao H, Roberts AP, Dekkers MJ, Liu X, Rohling EJ, Shi Z, et al. Late Miocene–Pliocene Asian monsoon intensification linked to Antarctic ice-sheet growth. Earth Planet Sci Lett. 2016;444:75–87.
Passey BH, Ayliffe LK, Kaakinen A, Zhang Z, Eronen JT, Zhu Y, et al. Strengthened East Asian summer monsoons during a period of high-latitude warmth? Isotopic evidence from Mio-Pliocene fossil mammals and soil carbonates from northern China. Earth Planet Sci Lett. 2009;277(3):443–452.
Wang ZH, Fang JY, Tang ZY, Lin X. Patterns, determinants and models of woody plant diversity in China. Proc Royal Soc B. 2011;278(1715):2122–2132.
Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15.
Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016;45(4):e18.
Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, et al. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45(W1):W6–W11.
Lohse M, Drechsel O, Kahlau S, Bock R. OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41(W1):W575–W581.
National Center for Biotechnology Information. https://www.ncbi.nlm.nih.gov/. Accessed 10 Oct 2020.
Jin J-J. PersonalUtilities. 2019. https://github.com/Kinggerm/PersonalUtilities. Accessed 15 Oct 2019.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780.
Degnan JH. Modeling hybridization under the network multispecies coalescent. Syst Biol. 2018;67(5):786–799.
Borowiec ML. AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ. 2016;4:e1660.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98.
Gonçalves DJP, Simpson BB, Ortiz EM, Shimizu GH, Jansen RK. Incongruence between gene trees and species trees and phylogenetic signal variation in plastid genes. Mol Phylogenet Evol. 2019;138:219–232.
Chernomor O, von Haeseler A, Minh BQ. Terrace aware data structure for phylogenomic inference from supermatrices. Syst Biol. 2016;65(6):997–1008.
R: A language and environment for statistical computing. http://www.R-project.org/. Accessed 12 Dec 2019.
Steenwyk JL, Buida TJ, III, Li Y, Shen X-X, Rokas A. ClipKIT: a multiple sequence alignment trimming software for accurate phylogenomic inference. PLoS Biol. 2020;18(12):e3001007.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14(6):587–589.
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: Improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35(2):518–522.
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–321.
Smith SA, Brown JW, Walker JF. So many genes, so little time: A practical approach to divergence-time estimation in the genomic era. PLoS One. 2018;13(5):e0197433.
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst Biol. 2018;67(5):901–904.
FigTree. http://tree.bio.ed.ac.uk/software/figtree/. Accessed 25 Nov 2018.
Nie Z-L, Wen J, Sun H. Phylogeny and biogeography of Sassafras (Lauraceae) disjunct between eastern Asia and eastern North America. Plant Syst Evol. 2007;267(1):191–203.
Li L, Madriñán S, Li J. Phylogeny and biogeography of Caryodaphnopsis (Lauraceae) inferred from low-copy nuclear gene and ITS sequences. Taxon. 2016;65(3):433–443.
Eklund H. Lauraceous flowers from the Late Cretaceous of North Carolina, U.S.A. Bot J Linn Soc. 2000;132(4):397–428.
Atkinson BA, Stockey RA, Rothwell GW, Mindell RA, Bolton MJ. Lauraceous flowers from the Eocene of Vancouver Island: Tinaflora beardiae gen. et sp. nov. (Lauraceae). Int J Plant Sci. 2015;176(6):567–585.
Li H, Liu B, Davis CC, Yang Y. Plastome phylogenomics, systematics, and divergence time estimation of the Beilschmiedia group (Lauraceae). Mol Phylogenet Evol. 2020;151:106901.
Li J, Qiu J, Liao W, Jin J. Eocene fossil Alseodaphne from Hainan Island of China and its paleoclimatic implications. Sci China Ser D: Earth Sci. 2009;52(10):1537.
Tang B, Han M, Xu Q, Jin J. Leaf cuticle microstructure of Machilus maomingensis sp. nov. (Lauraceae) from the Eocene of the Maoming basin, south China. Acta Geol Sin - Engl. 2016;90(5):1561–1571.
Bannister JM, Conran JG, Lee DE. Lauraceae from rainforest surrounding an early Miocene maar lake, Otago, southern New Zealand. Rev Palaeobot Palynol. 2012;178:13–34.
Morlon H, Lewitus E, Condamine FL, Manceau M, Clavel J, Drury J. RPANDA: an R package for macroevolutionary analyses on phylogenetic trees. Methods Ecol Evol. 2016;7(5):589–597.
Jetz W, Thomas GH, Joy JB, Hartmann K, Mooers AO. The global diversity of birds in space and time. Nature. 2012;491(7424):444–448.
Paradis E, Claude J, Strimmer K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 2004;20(2):289–290.
Rabosky DL, Grundler M, Anderson C, Title P, Shi JJ, Brown JW, et al. BAMMtools: an R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol Evol. 2014;5(7):701–707.
Plummer M, Best N, Cowles K, Vines K. CODA: convergence diagnosis and output analysis for MCMC. R news. 2006;6:7–11.
Zachos JC, Dickens GR, Zeebe RE. An early Cenozoic perspective on greenhouse warming and carbon-cycle dynamics. Nature. 2008;451(7176):279–283.
Höhna S, May MR, Moore BR. TESS: an R package for efficiently simulating phylogenetic trees and performing Bayesian inference of lineage diversification rates. Bioinformatics. 2016;32(5):789–791.
Höhna S, May MR, Moore BR. Phylogeny simulation and diversification rate analysis with TESS. 2015. https://cran.r-project.org/web/packages/TESS/vignettes/Bayesian_Diversification_Rate_Analysis.pdf. Accessed 15 Oct 2020.
National Specimen Information Infrastructure. http://www.nsii.org.cn/. Accessed 5 Dec 2020.
Barbour MG, Billings WD. North American terrestrial vegetation. Cambridge: Cambridge University Press; 2000.
Harris AJ, Wen J, Xiang Q-Y. Inferring the biogeographic origins of inter-continental disjunct endemics using a Bayes-DIVA approach. J Syst Evol. 2013;51(2):117–133.
Revell LJ. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol. 2012;3(2):217–223.
Pennell MW, Eastman JM, Slater GJ, Brown JW, Uyeda JC, FitzJohn RG, et al. geiger v2.0: an expanded suite of methods for fitting macroevolutionary models to phylogenetic trees. Bioinformatics. 2014;30(15):2216–2218.
Yu G, Smith DK, Zhu H, Guan Y, Lam TT-Y. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8(1):28–36.
Xiao T-W, Ge X-J. Plastomes of tribe Perseeae (Lauraceae). Science Data Bank. 2021. https://doi.org/10.11922/sciencedb.01280.
Acknowledgements
The authors thank Yu-Ying Zhou for DNA extraction, Tong-Jian Liu, Xiang-Qin Yu and Wei Wang for their data analysis and discussion advices. They also thank Yu Song for sharing plastid genomes from Lauraceae Chloroplast Genome Database (LCGDB; https://lcgdb.wordpress.com/). The authors would like to thank TopEdit (www.topeditsci.com) and AJ Harris for linguistic assistance during preparation of this manuscript.
Funding
The funders had no role in the material collection, experiment design, data analysis, decision to publish, or manuscript preparation. The authors acknowledge the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDB31000000.
Author information
Authors and Affiliations
Contributions
XJG and TWX conceived and designed the experiments; TWX assembled and annotated the plastid genomes, analyzed the data, and wrote the manuscript. HFY and XJG revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No specific permits were required, materials collection and molecular experiments were carried on following current Chinese regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Complete plastid genome map of tribe Perseeae. Different genes are color coded.
Additional file 2: Fig. S2.
ML tree inferred from IQ-TREE based on unpartitioned protein coding genes (PCG). The support values of Shimodaira-Hasegawa-like approximate likelihood ratio test (SH-aLRT; on the left) and ultrafast bootstrap (UFBS; on the right) are shown on the branches, respectively.
Additional file 3: Fig. S3.
ML tree inferred from IQ-TREE based on unpartitioned non-protein coding genes (NPCG). The support values of SH-aLRT (on the left) and UFBS (on the right) are shown on the branches.
Additional file 4: Fig. S4.
ML tree inferred from IQ-TREE based on partitioned genes of complete plastomes (CP). The support values of SH-aLRT (on the left) and UFBS (on the right) are shown on the branches.
Additional file 5: Fig. S5.
ML tree inferred from IQ-TREE based on partitioned PCG. The support values of SH-aLRT (on the left) and UFBS (on the right) are shown on the branches.
Additional file 6: Fig. S6.
ML tree inferred from IQ-TREE based on partitioned NPCG. The support values of SH-aLRT (on the left) and UFBS (on the right) are shown on the branches.
Additional file 7: Fig. S7.
Saturation indices for 197 loci shown as density plots. (a) Slopes of the linear regression between patristic and uncorrected pairwise distances. (b) R2 of the linear regression between patristic and uncorrected pairwise distances. The vertical and dashed lines indicate starting shoulder value. Loci that may be saturated are colored in red; the numbers of saturated loci are indicated aside.
Additional file 8: Fig. S8.
ML tree inferred from IQ-TREE based on unpartitioned CP-slope. CP-slope refers to CP after excluding saturated loci based on slopes of the linear regression. The support values of SH-aLRT (on the left) and UFBS (on the right) are shown on the branches, respectively.
Additional file 9: Fig. S9.
ML tree inferred from IQ-TREE based on unpartitioned CP-R2. CP-R2 refers to CP after excluding saturated loci based on R2 of the linear regression. The support values of SH-aLRT (on the left) and UFBS (on the right) are shown on the branches.
Additional file 10: Fig. S10.
Number of gaps in each locus. The locus with the highest number of gaps (ccsA-trnLUAG) is marked.
Additional file 11: Fig. S11.
Divergence times inferred from BEAST2. The red pentagrams refer to the fossil and secondary calibration points. The white numbers in black circles correspond to those in Fig. 2 and mentioned in the main text. The numbers and blue bars at nodes are divergence times before present and corresponding time intervals in the 95% highest posterior density (HPD).
Additional file 12: Fig. S12.
Credible shift set inferred from BAMM analysis. The values of f indicate the probabilities of speciation rate shifts in the maximum sampled posterior configuration.
Additional file 13: Fig. S13.
Speciation rate and location of rate shift when global sampling probability was 0.0675.
Additional file 14: Fig. S14.
Diversification rate inferred from CoMET in TESS. The shaded areas in (a) and (c) indicate 95% confidence intervals of speciation and extinction rates. 2lnBF (the heights of bars) higher than 6 indicate significant speciation rate shift, extinction rate shift, or mass extinction in (b), (d), and (e).
Additional file 15: Fig. S15.
The single-chain MCMC diagnostics for a CoMET analysis. The blue bars and dots indicate passed tests, while red bars and dots refer to failed convergence.
Additional file 16: Table S1.
The species names of tribe Perseeae.
Additional file 17: Table S2.
Characteristics of the 242 loci of tribe Perseeae and outgroups.
Additional file 19: Table S4.
The vegetation types of 52 sampled species in tribe Perseeae. Tropical forests, subtropical EBLFs, southeastern evergreen forests in North America are coded as 0, 1, and 2, respectively. Species distributed in both tropical forests and subtropical EBLFs is coded as 3.
Additional file 20: Table S5.
Model selection results inferred from geiger R package. AIC, Akaike Information Criterion.
Additional file 21: Table S6.
The newly sequenced species of tribe Perseeae and outgroups.
Additional file 22: Table S7.
The downloaded plastomes of Lauraceae.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xiao, TW., Yan, HF. & Ge, XJ. Plastid phylogenomics of tribe Perseeae (Lauraceae) yields insights into the evolution of East Asian subtropical evergreen broad-leaved forests. BMC Plant Biol 22, 32 (2022). https://doi.org/10.1186/s12870-021-03413-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-021-03413-8