
Abstract
Vicia amoena is a high-nutritional quality forage similar to alfalfa. However, studies on the genetic background of V. amoena are scarce. In the present study, the genetic variation of 24 V. amoena populations was assessed with newly developed simple sequence repeat (SSR) markers. A total of 8799 SSRs were identified in the V. amoena genomic-enriched sequences, and the most abundant repeat number was four. A total of 569 sampled individuals were assayed to evaluate the genetic diversity of the V. amoena populations based on 21 polymorphic SSR primers. The polymorphism information content (PIC) ranged from 0.896 to 0.968, with an average of 0.931, which indicated that the markers were highly informative. Based on analysis of molecular variance, 88% of the variance occurred within populations, and the remaining 12% of the variance occurred among populations. The high degree of gene flow (Nm= 4.958) also showed slight differentiation among the V. amoena populations. The V. amoena populations were mainly clustered by steppe and mountain habitats based on principal coordinate analysis (PCoA) and STRUCTURE analysis. This indicated that the elevation and special habitat of geographical origins may be important factors affecting the clustered pattern of V. amoena populations. Neighbour-joining (NJ) analysis did not separate the populations well by geographical origin, which indicated that the genetic structure of V. amoena was complex and needs further study. Overall, our results showed that the newly developed SSR markers could benefit the V. amoena research community by providing genetic background information to help establish a foundation for breeding improvement and germplasm resource conservation.
Similar content being viewed by others
Background
Vicia amoena is an herbaceous, allotetraploid (2n=24), perennial legume species native to Eastern Asia (Siberia, Mongolia, China, Japan, and Korea) that is especially widely dispersed in northern China [1, 2]. It has high nutritional quality, strong abiotic stress tolerance, and wide adaptability. The protein content and the amino acid content of V. amoena are comparable to those of alfalfa (Medicago sativa) [3]. Moreover, V. amoena is also used as a traditional Chinese medicinal herb to treat oedema, rheumatoid arthritis and contracture [4]. However, genetic research on this important forage legume is scarce, with most researchers instead focusing on its chemical components. Unravelling the genetic diversity and population structure of V. amoena is very important for understanding its genetic background, which is a prerequisite for future genetic research, breeding programme development and genetic resource conservation.
Microsatellites or simple sequence repeat (SSR) markers are a powerful molecular method for quantifying genetic variation in plants due to their high polymorphism [5]. SSR markers are characterized by repeated sequences comprising mono-, di-, tri-, tetra-, penta- or hexa-nucleotide units that are characterized by tandem repeats (1-10 nucleotide motifs) that exhibit locus-specific codominance and high heterozygosity, are distributed throughout the genome, and are easier to detect than other molecular markers [6]. Microsatellite markers have been successfully used in the assessment of many plants, e.g., Vicia faba [7, 8], Campomanesia adamantium [9], Populus deltoides [10], Olea europaea [11], and Cunninghamia lanceolata [12].
Overall, SSRs are one of the most informative molecular markers for plant genetic research, but the isolation of SSR markers traditionally based on probe hybridization is an experimentally demanding, labour-intensive, and economically costly process [13]. Advancements in sequencing and bioinformatic analysis techniques have provided good opportunities for generating new SSR markers. For example, next-generation sequencing (NGS) technology is a powerful tool that can be used for fast and cost-effective SSR discovery [14, 15]. To date, a large number of SSR markers have been developed by high-throughput sequencing in many plants, such as Medicago sativa [16], Vicia sativa [15], Elymus sibiricus [17], Onobrychis viciifolia [18], Angelica gigas [19], Lentinula edodes [20], and Spondias tuberosa [21].
In the present study, we developed SSR markers using the HiSeq 4000 PE150 sequencing platform. We then used 21 polymorphic pairs to analyse the genetic diversity and population structure of 24 V. amoena populations (569 total individuals) in China, which may support studies on molecular diversity and breeding programmes. Our goals are (1) to assess the validity of these newly developed SSR markers and (2) to obtain an accurate representation of the genetic diversity and population structure of V. amoena.
Material and methods
Plant materials and DNA isolation
A total of 569 individuals from 24 sites throughout the natural distribution of V. amoena in China were collected in the present study (Table 1). Of these individuals, 281 individuals from 13 populations were collected in the field. The other 288 individuals from 11 sites were obtained from seeds provided by the National Herbage Germplasm Conservation Centre of China (Beijing, China). Genomic DNA was extracted from fresh or silica gel-dried leaf tissues using a Plant Genomic DNA Extraction Kit (Tiangen, Beijing, China) according to the manufacturer’s protocol.
SSR marker detection, identification, and primer design
An Illumina paired-end library was constructed by the NEBNext® Ultra™ II DNA Library Preparation Kit (New England Biolabs (Beijing) Ltd., China) and sequenced on the Illumina HiSeq 4000 PE150 sequencing platform. Approximately 17.5 Gb of raw data was generated, and the raw sequence reads were filtered for primer/adaptor sequences and low-quality reads with the NGS QC Tool Kit [22]. Sequencing reads were assembled using SPAdes 3.6.1 software [23] with the parameter Kmer=95, and 198,659 contigs were finally obtained.
MISA software [24, 25] was used to identify unique reads containing microsatellite repeats. The search was performed for a minimum repeat number of 5, 4, 3, 3 and 3 for di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. Primers were designed on the basis of flanking sequences of SSR microsatellite loci by using Primer 3. The parameters of primer design were set as follows: the primer size was between 18 and 25 bp with an optimal size of 22 bp, the annealing temperature was between 55 and 65 °C with the optimal temperature of 60 °C, the PCR product size was between 80 and 300 bp, and default values were selected for other settings.
M13-SSR PCR amplification
Twenty-one SSRs were selected through a preliminary experiment, and this number of markers was suitable for evaluating plant genetic diversity [11, 26, 27]. Twenty-one primer pairs (Table 2) that successfully amplified fragments in the 569 individuals were further characterized for polymorphisms using the M13-SSR PCR protocol. There were three primers in the M13-SSR PCR system: a forward primer, a reverse primer with an M13-tail (5’-CACGACGTTGTAAAACGAC-3’) at the 5’ end, and a fluorescently labelled M13 universal primer. The first two primers were synthesized by Sangon Biotech (Shanghai, China) Co., Ltd., and the third primer was synthesized by Thermo Fisher Scientific (Shanghai, China). The four fluorescently labelled primers were FAM, NED, VIC, and ROX.
The total volume of the PCR was 10 μL, including 1 μL genomic DNA (30 ng), 0.5 μL forward M13 primer, 0.5 μL reverse primer, 0.5 μL fluorescent M13 primer, 1 μL 10× buffer, 1 μL dNTP, 0.1 μL Taq enzyme, and 5.4 μL ddH2O. The PCR amplification procedure was as follows: 3 min at 94 °C, followed by 30 cycles of 30 s at 94 °C, 30 s at 60 °C, and 30 s at 72 °C, with a final extension at 72°C for 10 min. The PCR products were subsequently detected by an ABI 3730xl Genetic Analyzer Sequencer at Sangon Biotech (Shanghai, China) Co., Ltd., and the outputs were analysed using the software GeneMarker v2.2.0 (SoftGenetics, State College, Pennsylvania, USA).
Data analysis
The number of alleles (Na), the number of effective alleles (Ne), Shannon’s information index (I), the observed heterozygosity (Ho), the expected heterozygosity (He), and the percentage of polymorphic loci (PPL) were determined to evaluate the genetic diversity of the SSRs and V. amoena populations. The genetic differentiation index (Fst) and genetic distance were calculated and principal coordinate analysis (PCoA) and analysis of molecular variance (AMOVA) were performed by GenAlEx 6.5 [28]. A NJ tree was constructed using MEGA X software [29]. Population genetic structure was determined using the model-based program in STRUCTURE 2.3.4 software with a Bayesian approach [30, 31]. The most likely number of populations (K) was identified among 2-24, and 10 interactions were performed for each value of K. The length of burn-in Markov chain Monte Carlo (MCMC) replications was set to 500,000, followed by 100,000 MCMC replications in each run. The optimal K capturing the major structure in the V. amoena data was determined using Structure Harvester (http://taylor0.biology.ucla.edu/structureHarvester/) [32, 33]. All tetraploid genotype data were converted into binary data using the POLYSAT v1.2 package in R [34]. Polymorphic information content (PIC) was calculated using the formula PIC = 1-∑Pi2, where Pi is the frequency of the i-th allele [35].
Results
Characterization of the developed SSR markers
A total of 8799 SSRs with 3 or more di-, tri-, tetra-, penta-, and hexa-nucleotide repeat units were identified in the enriched sequences of the V. amoena genome. The sequencing data generated in the present study have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (PRJNA742214). Among the SSRs, 2089, 3878, 2055, 533, and 244 were di-, tri-, tetra-, penta-, and hexa-nucleotides SSRs, respectively (Table 3). Of the dinucleotide motifs, AT/TA (25.64%) was the most abundant, followed by AG/TC (20.84%), AC/TG (20.44%), CA/GT (18.60%), and CT/GA (13.58%). The CG/GC motif was the least frequent (0.90%) dinucleotide. Of the trinucleotide motifs, AAC/TTG (14.75%) was the most abundant, followed by CAA/GTT (14.13%), ACA/TGT (13.54%), AAG/TTC (6.13%) and AGA/TCT (6.06%). The remaining trinucleotide motifs were present in less than 5% of the total. Of the tetra-, penta- and hexa-nucleotide motifs, CATA/GTAT (5.89%), ATAAT/TATTA (2.81%), and AAAAAG/TTTTTC (5.73%) were the most abundant, respectively (Table S1). The average length (bp) of di-, tri-, tetra-, penta- and hexa-nucleotide SSRs was 12.67, 13.25, 12.64, 15.87 and 19.30, respectively. The repeat number of 3035 SSR motifs (34.49%) was four, the repeat number of 2601 SSRs (29.56%) was three, and the repeat number of 2026 SSRs (23.03%) was five. The repeat numbers of 551 (6.26%), 199 (2.26%), and 142 (1.61%) SSRs were six, seven, and eight, respectively. The distribution frequency of the other 245 SSRs was less than 1% (Table 4).
For the 21 polymorphic SSR primers, the average allele number (Na) was 3.533, ranging from 3.250 to 15.542 (Table 5). The number of effective alleles (Ne) ranged from 2.680 to 9.751, with an average of 2.521 alleles. Shannon’s information index (I) ranged from 0.999 to 2.655, with an average of 0.930. The average observed heterozygosity (Ho) value was 0.713, ranging from 1.000 to 2.000. The expected heterozygosity (He) values ranged from 0.642 to 1.551, with an average of 0.485. The Fst ranged from 0.029 to 0.140, with an average of 0.384, and the average value of Nm was 2.119, ranging from 1.538 to 8.380. Meanwhile, the PIC ranged from 0.896 to 0.968, with an average of 0.931.
Genetic diversity and structure of V. amoena populations
The genetic diversity of the 24 V. amoena populations (n=569) was also assessed, revealing high mean per-population estimates of allele and genetic diversity (Na=3.533; Ne=2.521; I=0.930; Ho=0.713; He=0.485; Table 6). The PPL of the 24 V. amoena populations ranged from 54.76% to 83.33%, with an average of 71.33%. The genetic diversity was highest in ZX1141 (Na=4.976; Ne=3.191; I=1.159; Ho=0.810; He=0.570) and lowest in QHA (Na=1.857; Ne=1.526; I=0.583; Ho=0.548; He=0.340). The same pattern was found for PPL, which was also higher in ZX1141 (80.95%) than in QHA (54.76%).
In addition, AMOVA was conducted to evaluate variance components among V. amoena populations (Table 7). The results revealed significant differences within and among populations (P=0.001). A much greater proportion of the variance was observed within populations (88%), than among populations (12%). Population differentiation (Fst=0.048) was significant (P=0.001), and the Nm was 4.958.
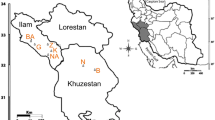
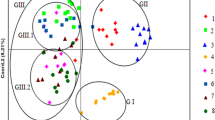
PCoA, NJ tree construction, and STRUCTURE analysis were conducted to further evaluate the genetic relationships among the 24 V. amoena populations. In the PCoA (Fig. 1), the QHA population was separated into a single cluster. The 23 populations were clustered into cluster A and cluster B. Cluster A included the populations from Inner Mongolia (YHT, N50, NM03, ZQ, M99, B514, B515, and B516), Shanxi (YX and ZX476), Heilongjiang (ZD), and Beijing (ZX986 and ZX1141). The other 10 populations were clustered together, including those from Inner Mongolia (STG, YDZ, XLT, and MQ), Heilongjiang (HEB), Shanxi (SJ and ZX562), Beijing (ZX541), Hebei (ZX987), and Qinghai (QHB).

PCoA of the 24 V. amoena populations
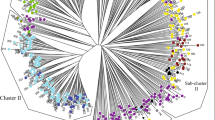
Additionally, an NJ tree of V. amoena individuals was constructed based on Nei’s genetic distance, and five clusters (Clusters I, II, III, IV, and V) were identified (Fig. 2). Cluster I included the populations from Inner Mongolia (MQ, STG, ZQ, XLT, B514, and B515), Shanxi (YX and SJ), Heilongjiang (HEB), and Beijing (ZX541). Cluster II could be further separated into three sub-clusters: one sub-cluster included the populations from Qinghai (QHA and QHB) and Inner Mongolia (YDZ), the individuals of M99 (Inner Mongolia) formed another sub-cluster, and the individuals of ZX562 (Shanxi) formed the third sub-cluster. ZD (Heilongjiang); ZX987 (Hebei); ZX476 and ZX562 (Shanxi); NM03, B514, B515, and B516 (Inner Mongolia); and ZX541, ZX986, and ZX1141 (Beijing) formed Cluster III. Some individuals from Inner Mongolia (B514, B515, B516, YDZ, MQ, N50, ZD, XLT, and STG), Shanxi (ZX476 and YX), Beijing (ZX986 and ZX1141), Hebei (ZX987), and Heilongjiang (HEB) formed Cluster IV. Cluster V included other individuals from Qinghai (QHA and QHB), Inner Mongolia (B515, B516, XLT, YHT, and ZQ), Shanxi (SJ and ZX476), and Beijing (ZX1141).

NJ analysis of 24 V. amoena populations based on SSR datasets (Cluster I, red; Cluster II, green; Cluster III, blue; Cluster IV, yellow; Cluster V, pink)
For the entire SSR dataset (24 populations, n=569), STRUCTURE analysis yielded the highest likelihood when samples were clustered into 10 groups (K=10, Fig. S1). The populations from Heilongjiang were assigned to cluster 1 (HEB, 44.9%) and cluster 2 (ZD, 74.1%). The populations from Inner Mongolia were assigned to 7 clusters, and the individuals of B514 (73.3%) formed a separate cluster (cluster 10). Only populations YDZ (75%) and YHT (94.1%) were assigned to cluster 4. The other populations were clustered with populations from other regions: N50 (87.5%) and MQ (53%) were assigned to cluster 1; XLT (77.4%) was assigned to cluster 2; M99 (44%), STG (63.5%), and ZQ (61.4%) were assigned to cluster 3; NM03 (73.8%) and B515 (57.5%) were assigned to cluster 5; and ZX541 (45.5%) was clustered into cluster 7. The Beijing populations were all assigned to cluster 5, which included ZX541 (44%), ZX986 (72.1%), and ZX1141 (27.8%). The individuals of ZX1141 (32.7%) were assigned to cluster 3. The individuals of Hebei (ZX987, 73.5%) were assigned to cluster 5 with the Beijing populations. Almost all the individuals of ZX562 (93.9%) formed a separate cluster (cluster 6), and the individuals of YX (84.1%) formed a separate cluster (cluster 9). The other two populations from Shanxi were assigned to cluster 7 (SJ, 63.8%) and cluster 8 (ZX476, 67.8%). The populations from Qinghai were mainly assigned to cluster 7 (QHB, 75.6%) and cluster 8 (QHA 96.1%) (Fig. 3, Table S2).

STRUCTURE analysis of the entire SSR dataset (24 populations, n=569). a The geographic distribution of the 24 V. amoena populations inferred with K=10. The different colours of the pie charts represent the proportions of the populations in the 10 clusters (Table S2). b STRUCTURE output with K=10 (Fig. S1) showing the population structure among 569 individuals; vertical lines represent individuals.
Discussion
The development and polymorphism of SSR markers
Genetic research on V. amoena has developed slowly due to a lack of sufficient genetic information and effective molecular marker systems. SSRs are one of the most important marker systems for plant genetic studies with genetic diversity evaluation, marker-assisted selection (MAS) breeding, quantitative trait locus (QTL) mapping, and variety identification and are extensively distributed throughout eukaryotic genomes [36, 37]. However, traditional SSR development methods are labour intensive [13]. At present, SSR markers developed by high-throughput sequencing are reliable and effective [19, 38,39,40,41]. Genomic SSRs have not been developed thus far in V. amoena, and a new set of highly polymorphic SSR markers was successfully developed in the present study. A total of 8799 SSRs were developed in V. amoena at the genome-wide scale, which was far greater than the 1071 EST-SSRs developed by transcriptome sequencing in V. sativa [15]. Our work provides a powerful tool for genetic research on V. amoena in future breeding programmes and resource conservation. Among the SSR markers, trinucleotide repeats were the most abundant (44.07%) type, similar to the relative proportions of EST-SSR motif types observed in V. sativa [15] and Medicago sativa [16]. The results indicated that the trinucleotide SSRs in the V. amoena genome are mainly located in exon regions. The frequent distribution of trinucleotide repeats in coding regions indicates the effects of selection and evolution [41].
The 21 SSR markers used in this study offered an informative and applicable approach for the evaluation of genetic relationships among the V. amoena populations. The genetic diversity parameter values indicated the high polymorphism of the 21 SSR markers. The observed heterozygosity (Ho) and expected heterozygosity (He) values also revealed a high degree of genetic variability among the V. amoena populations [11]. The values of PIC, Ho and He were all higher than those of the EST-SSRs reported in V. sativa [15]. This could be related to the different methods of SSR marker development and the different genetic backgrounds of various plant species.
Genetic differentiation and genetic structure of V. amoena populations
In the present study, a high level of genetic diversity (I=0.930) was detected among the V. amoena populations by the newly developed SSR markers. This genetic diversity was more evident than that detected by SRAP and ISSR markers in a previous report (I=0.397) [2]. Two reasons for this difference are that SSR markers are more effective than the other two types of markers [42] and more natural populations were examined in the present study. Among the populations, those from Qinghai Province showed a lower level of genetic diversity, which may be due to their unique geographical location on the Qinghai-Tibet Plateau. The populations from tall mountain areas with high forest coverage at approximately 40°N had a higher level of genetic diversity. Genetic variation within the populations (88%) was higher than that among the populations (12%) in this study. The results were consistent with the characteristics of outcrossing species [43, 44], which can be attributed to allogamous reproductive behaviour. The variation in V. amoena mainly comes from intrapopulation variation, confirming that V. amoena is a cross-pollinating plant.
The 24 V. amoena populations could be separated into three clusters via PCoA. The populations were mainly separated by habitat, i.e., mountain meadow, Leymus chinensis steppe, and undergrowth on mountains. The results indicated that the elevation of the geographical origin may be an important factor explaining the clustered pattern of V. amoena and that special habitat is another important factor. Similar results were found in the STRUCTURE analysis. The inferred subpopulations were broadly separated based on the best K value (K=10). The populations were mainly clustered among Leymus chinensis steppe, mountain areas with high forest coverage, and the Qinghai-Tibet Plateau. The results showed that the clusters of V. amoena were impacted by different landforms and the special topography of the Qinghai-Tibet Plateau. It would be worth exploring how the special topography affects the genetic differentiation of V. amoena in the future.
Additionally, the NJ analysis of V. amoena based on the entire SSR dataset revealed five major groups and showed an interesting pattern. The individuals from the populations on mountains were clustered with the populations from the Qinghai-Tibet Plateau. The other populations from the mountains and Leymus chinensis steppe were gathered in three clusters. The clustered pattern in the NJ analysis did not show clear boundaries among the different habitats and elevations. The high gene flow (Nm= 4.958) also weakened the differentiation among the V. amoena populations. The results indicated that the genetic structure of V. amoena populations was complex and affected by many factors, which needs further analysis. This might be due to the special climatic conditions, habitats, and geomorphic conditions [2].
In conclusion, our results confirmed that the V. amoena populations in China contained a high level of genetic diversity. There is a tendency for the genetic structure of the populations to be correlated with geographical origin and comprehensive environmental factors. Our findings and the SSRs newly developed in the present study provide a strong tool for breeding improvement and germplasm resource conservation in V. amoena.
Availability of data and materials
The data supporting this article are included within the article and its additional files. The original sequencing data generated in the study have been deposited into the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA742214).
Abbreviations
- SSR:
-
Simple sequence repeat
- PIC:
-
Polymorphism information content
- Na:
-
Number of alleles
- Ne:
-
Number of effective alleles
- I:
-
Shannon’s information index
- Ho:
-
Observed heterozygosity
- He:
-
Expected heterozygosity
- PPL:
-
Percentage of polymorphic loci
- AMOVA:
-
Analysis of molecular variance
- UPGMA:
-
Unweighted pair-group method with arithmetic means
References
Li R, Taylor S, Jenkins G. Unravelling the phylogeny of tetraploid Vicia amoena (Fabaceae) and its diploid relatives using chromosomal landmarks. Hereditas. 2001;134(3):219–24.
Liu Y. Zhang J-m, Wang X-g, Liu F, Shen Z-b: Genetic diversity in Vicia amoena (Fabaceae) germplasm resource in China using SRAP and ISSR markers. Biochem Syst Ecol. 2013;51:86–93.
Li Z, Ma M, Zhao W, Li C. Nutritional dynamics at different growth stages of Vicia amoena and its effect on rumen fermentation and methane production in vitro. Feed Res. 2016;000(002):1–6.
Yang QH, Luo SQ, Liu LL, Huo HY. Preliminary test of chemical components for Vicia amoena, a Mongolian medicine. Acta Academiae Medicinae Nmongol. 2010;132(5):475–7.
Zhang J, Chen T, Wang J, Chen Q, Luo Y, Zhang Y, et al. Genetic diversity and population structure in cherry (Cerasus pseudocerasus (Lindl). G. Don) along Longmenshan Fault Zones in China with newly developed SSR markers. Scientia Horticult. 2016;212:11–9.
Vieira ML, Santini L, Diniz AL, Munhoz Cde F. Microsatellite markers: what they mean and why they are so useful. Genet Mol Biol. 2016;39(3):312–28.
El-Esawi MA. SSR analysis of genetic diversity and structure of the germplasm of faba bean (Vicia faba L.). Comptes Rendus Biol. 2017;340(11-12):474–80.
Gol S, Doganlar S, Frary A: Relationship between geographical origin, seed size and genetic diversity in faba bean (Vicia faba L.) as revealed by SSR markers. Mol Genet Genomics. 2017;292(5):991–9.
Crispim BDA, Déo TG, Fernandes JDS, de Vasconcelos AA, Vieira MDC, Carnevali TO, et al. Development and characterization of microsatellite markers in Campomanesia adamantium, a native plant of the Cerrado ecoregions of South America. Appl Plant Sci. 2019;7(9):e11287.
Chen C, Chu Y, Ding C, Su X, Huang Q. Genetic diversity and population structure of black cottonwood (Populus deltoides) revealed using simple sequence repeat markers. BMC Genet. 2020;21(1):2.
Li D, Long C, Pang X, Ning D, Wu T, Dong M, et al. The newly developed genomic-SSR markers uncover the genetic characteristics and relationships of olive accessions. PeerJ. 2020;8:e8573.
Lin E, Zhuang H, Yu J, Liu X, Huang H, Zhu M, et al. Genome survey of Chinese fir (Cunninghamia lanceolata): Identification of genomic SSRs and demonstration of their utility in genetic diversity analysis. Sci Rep. 2020;10(1):4698.
Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, et al. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot. 2012;99(2):193–208.
Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity (Edinb). 2011;107(1):1–15.
Liu Z, Liu P, Luo D, Liu W, Wang Y. Exploiting Illumina sequencing for the development of 95 novel polymorphic EST-SSR markers in common vetch (Vicia sativa subsp. sativa). Molecules. 2014;19(5):5777–89.
Liu Z, Chen T, Ma L, Zhao Z, Zhao PX, Nan Z, et al. Global transcriptome sequencing using the Illumina platform and the development of EST-SSR markers in autotetraploid alfalfa. PLoS One. 2013;8(12):e83549.
Zhou Q, Luo D, Ma L, Xie W, Wang Y, Wang Y, Liu Z. Development and cross-species transferability of EST-SSR markers in Siberian wildrye (Elymus sibiricus L.) using Illumina sequencing. Sci Rep. 2016;6:20549.
Kempf K, Mora-Ortiz M, Smith LM, Kolliker R, Skot L. Characterization of novel SSR markers in diverse sainfoin (Onobrychis viciifolia) germplasm. BMC Genet. 2016;17(1):124.
Gil J, Um Y, Kim S, Kim OT, Koo SC, Reddy CS, et al. Development of genome-wide ssr markers from angelica gigas nakai using next generation sequencing. Genes (Basel). 2017;8(10):238.
Lee HY, Moon S, Shim D, Hong CP, Lee Y, Koo CD, Chung JW, Ryu H. Development of 44 Novel polymorphic ssr markers for determination of shiitake mushroom (Lentinula edodes) cultivars. Genes (Basel). 2017;8(4):109.
Balbino E, Martins G, Morais S, Almeida C. Genome survey and development of 18 microsatellite markers to assess genetic diversity in Spondias tuberosa Arruda Câmara (Anacardiaceae) and cross-amplification in congeneric species. Mol Biol Rep. 2019;46(3):3511–7.
Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7(2):e30619.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Thiel T, Michalek W, Varshney RK, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 2003;106(3):411–22.
Beier S, Thiel T, Münch T, Scholz U, Mascher M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33(16):2583–5.
de Dato GD, Teani A, Mattioni C, Aravanopoulos F, Avramidou EV, Stojnic S, et al. Genetic Analysis by nuSSR Markers of Silver Birch (Betula pendula Roth) Populations in Their Southern European Distribution Range. Front Plant Sci. 2020;11:310.
Li B, Lin F, Huang P, Guo W, Zheng Y. Development of nuclear SSR and chloroplast genome markers in diverse Liriodendron chinense germplasm based on low-coverage whole genome sequencing. Biol Res. 2020;53(1):21.
Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics. 2012;28(19):2537–9.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 2018;35(6):1547–9.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–87.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mole Ecol Notes. 2007;7(4):574–8.
Earl DA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4(2):359–61.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–20.
Clark LV, Jasieniuk M. POLYSAT: an R package for polyploid microsatellite analysis. Mole Ecol Resour. 2011;11(3):562–6.
Powell W, Morgante M, Andre C, Hanafey M, Vogel J, Tingey S, et al. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol Breed. 1996;2(3):225–38.
Thakur O, Randhawa GS. Identification and characterization of SSR, SNP and InDel molecular markers from RNA-Seq data of guar (Cyamopsis tetragonoloba, L. Taub.) roots. BMC Genom. 2018;19(1):951.
Yang J, Zhang J, Han R, Zhang F, Mao A, Luo J, et al. Target SSR-Seq: a novel ssr genotyping technology associate with perfect ssrs in genetic analysis of cucumber varieties. Front Plant Sci. 2019;10:531.
Bazzo BR, de Carvalho LM, Carazzolle MF, Pereira GAG, Colombo CA. Development of novel EST-SSR markers in the macaúba palm (Acrocomia aculeata) using transcriptome sequencing and cross-species transferability in Arecaceae species. BMC Plant Biol. 2018;18(1):276.
Park S, Son S, Shin M, Fujii N, Hoshino T, Park S. Transcriptome-wide mining, characterization, and development of microsatellite markers in Lychnis kiusiana (Caryophyllaceae). BMC Plant Biol. 2019;19(1):14.
Taheri S, Abdullah TL, Rafii MY, Harikrishna JA, Werbrouck SPO, Teo CH, et al. De novo assembly of transcriptomes, mining, and development of novel EST-SSR markers in Curcuma alismatifolia (Zingiberaceae family) through Illumina sequencing. Sci Rep. 2019;9(1):3047.
Chai M, Ye H, Wang Z, Zhou Y, Wu J, Gao Y, et al. Genetic Divergence and Relationship Among Opisthopappus Species Identified by Development of EST-SSR Markers. Front Genet. 2020;11:177.
Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK. Microsatellite markers: an overview of the recent progress in plants. Euphytica. 2011;177(3):309–34.
Van Treuren R, Bas N, Goossens PJ, Jansen J, Van Soest LJ. Genetic diversity in perennial ryegrass and white clover among old Dutch grasslands as compared to cultivars and nature reserves. Mol Ecol. 2005;14(1):39–52.
George J, Dobrowolski MP, van Zijll de Jong E, Cogan NO, Smith KF, Forster JW. Assessment of genetic diversity in cultivars of white clover (Trifolium repens L.) detected by SSR polymorphisms. Genome. 2006;49(8):919–30.
Acknowledgments
Not applicable
Funding
The present work was financially supported by the National Natural Science Foundation of China (No. 31772657). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
FW and XW designed the experiments, performed the experiments, analyzed the data, prepared tables and the figures, and wrote the main manuscript text. SZ, QG, FL, and JW performed the experiments. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no conflicts of interest
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The repeats number of different SSR motifs. Table S2. The proportion of each population in the genetic structure analysis. Figure S1. The best K-value of the genetic structure based on STRUCTURE analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, F., Zhang, S., Gao, Q. et al. Genetic diversity and population structure analysis in a large collection of Vicia amoena in China with newly developed SSR markers. BMC Plant Biol 21, 544 (2021). https://doi.org/10.1186/s12870-021-03330-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-021-03330-w