
Abstract
Background
Papaya (Carica papaya L.) is a popular climacteric fruit, undergoing various physico-chemical changes during ripening. Although papaya is widely cultivated and consumed, few studies on the changes in metabolism during its ripening process at the proteasome level have been performed. Using a newly developed TMT-LCMS analysis, proteomes of papaya fruit at different ripening stages were investigated.
Results
In total, 3220 proteins were identified, of which 2818 proteins were quantified. The differential accumulated proteins (DAPs) exhibited various biological functions and diverse subcellular localizations. The KEGG enrichment analysis showed that various metabolic pathways were significantly altered, particularly in flavonoid and fatty acid metabolisms. The up-regulation of several flavonoid biosynthesis-related proteins may provide more raw materials for pigment biosynthesis, accelerating the color variation of papaya fruit. Variations in the fatty acid metabolism- and cell wall degradation-related proteins were investigated during the ripening process. Furthermore, the contents of several important fatty acids were determined, and increased unsaturated fatty acids may be associated with papaya fruit volatile formation.
Conclusions
Our data may give an intrinsic explanation of the variations in metabolism during the ripening process of papaya fruit.
Similar content being viewed by others


Background
Fruit is a mature ovary including carpel tissues, and several fleshy fruits are important components of human and animal diets [1]. Fruit ripening is a complex process defined by dramatic metabolisms and textural transformations, color changes, softening, nutritional ingredient accumulation and flavor compound production [1, 2]. Although softening increases the nutritional and flavor properties of fruit, it also leads to biomass loss and postharvest deterioration [3].
Fruit softening is a deep-studied process that involves cell wall degradation [4]. The cell walls of fruit are composed of pectins, poly-galacturonans, celluloses and hemicelluloses, and degradation of these components is the major cause of tissue softening during ripening [5]. The pectin matrix, composed of both linear and branched polysaccharides, mainly accumulates in the primary cell wall. Several specific enzymes, such as pectate lyase, polygalacturonase and pectin methylesterase, show essential roles in the hydrolysis of polysaccharide chains of the pectin matrix [5, 6].
For climacteric fruits, the softening phenomenon is also controlled by a complex series of non-enzymatic events [7, 8]. During fruit ripening, abundant flavor and nutritional metabolites significantly change, including sugars, organic acids, lipids and amino acids, leading to modification of fruit texture [9]. For instance, sugars, the major flavor and aroma components, increase rapidly apricot fruit during development and ripening process [10]. In starch-rich fruit, starch and organic acids are converted to sugar by a metabolic degradation pathway [11]. Additionally, lipid components of fruits are assumed to enhance the formation of aromas and flavors in most climacteric fruits [12]. The variations in the composition of fatty acids during the fruit ripening process have been revealed in different climacteric fruits [13]. For example, most fatty acids increased during the ripening of mango fruit [14].
Papaya (Carica papaya L.), a climacteric fruit widely planted and consumed, displays quick softening and a short shelf-life [15, 16]. Once the climacteric fruit is mature, its soft texture and susceptibility to pathogenic fungi make the storage life short [17]. To overcome the commercial trading problem and prolong the shelf life, many studies on papaya have been carried out [18]. In papaya fruit, the physico-chemical changes during ripening are affected by the expression of ripening-related genes. A previous transcriptome identified 414 ripening-related genes, including those of MADS-box, NAC and ERF families, providing molecular information on papaya [19]. Several years ago, a fruit-specific expressed subtilase gene was identified in papaya by rapid amplification of cDNA ends and PCR primer walking techniques [20]. Application of 1-MCP (1-methylcyclopropene) can block ethylene signaling and so improve the shelf life [21]. Expression analysis of ethylene signaling pathway-related genes showed a relationship between chilling injury and ethylene signaling in papaya [16]. Physiological degradation of pectin in papaya cell walls has also been studied. For example, up-regulated expression levels of genes for PG, endoxylanase and β-galactosidase (β-GAL) were positively correlated with postharvest papaya fruit ripening [22, 23]. However, molecular and enzymatic mechanisms underlying the softening of papaya fruit during ripening remain largely unknown.
Many studies on the physiological and genomic variations during the ripening process of papaya fruit have been done [19, 23, 24]. To date, several proteomic data sets of papaya have been published. Using the 2-DE approach, differential accumulated proteins responsive to 1-MCP treatment during ripening have been identified [25]. Another comparative proteomic analysis showed that certain proteins, such as enolase, esterase and ADH3, show an essential role in maturation of somatic papaya embryo cells [26]. Recently, a differential proteome of virus-infected pre-flowering C. papaya vs. uninfected plants was published [27]. In our study, we employed an integration of the basic HPLC fractionation and LC–MS/MS approach to identify a total of 2818 differential accumulated proteins (DAPs) of papaya fruit during ripening. Our results offer important information about how to expend the shelf life of postharvest papaya fruit.
Methods
Plant materials and sampling
Two-year-old papaya trees of cultivar ‘Sunrise’ were planted in a 4 m × 4 m arrangement. The seedlings of papaya tree were purchased by the Yixin horticulture company (Zhanjiang, China), who provided permission to use the seedlings for our scientific research. The experimental station was at Lingnan Normal University campus in Zhanjiang, China. Plant experiment was performed in the Life Science and Technology School, Lingnan Normal University, according to a plant protocol approved by the Research Ethics Committee of Lingnan Normal University. All papaya trees were fertilized twice a week with a standard nutrient solution. Two fruit groups at similar color break stage (5% ≤ peel color ≤10% yellow) were harvested. The fruit coloring index was defined and calculated as follows: \( \mathrm{coloring}\ \mathrm{index}=\sum \frac{\left(\mathrm{coloring}\ \mathrm{grade}\times \mathrm{number}\ \mathrm{of}\ \mathrm{fruit}\right)}{\mathrm{total}\ \mathrm{number}\ \mathrm{of}\ \mathrm{fruit}}. \)
There are three papaya in one sample, and three samples for one group. The selected fruit was cleaned with deionized H2O, and then dipped into 0.2% of hypochloride solution for 15 min to eliminate potential microbes in growth chamber of our lab. The conditions of the chamber were set as: temperature of 26 ± 1 °C, a light/dark cycle of 16/8 h and humidity of 65–70%.
Physiological parameter measurements
Measurements of the physiological parameters of papaya fruit were performed according to the previous study [15]. Three biological replicate were used. Briefly, the firmness of fruit was determined using a GY-J mini fruit tester (Top Instrument Co Ltd., Shanghai, China). For total soluble solid (TSS) measurement, 5.0 g of fruit pulp from three independent fruit was crushed manually with a mortar and pestle to extract the juice. The TSS (%) was measures using a J1-3A refractometer (Guangdong Scientific Instruments, Guangzhou, China). The same juice was titrated against 0.1 N NaOH until a faint pink color was obtained at the end point and titratable acidity was expressed as percentage citric acid. For respiration rate determination, three independent papaya fruit from each group were weighed and sealed in 2 L containers at 25 °C. The concentration of CO2 in each container was measured using an infrared Li-6262 CO2/H2O gas analyzer (LI-COR, USA).
Observation by light microscopy and scanning electron microscopy
Fragments (0.5 cm long) of surface tissues were collected from whole fruit at different ripening stages. Samples were fixed in formalin acetic acid (FAA)50 for 48 h and transferred into 70% ethanol. The tissue samples were dehydrated in several varied concentrations of ethanol, and then embedded in wax. Next, fruit samples were sliced into a number of 12-μm thick sections using a RM2255 rotary microtome (Leica, Wetzlar, Germany) and spreaded on glass slides. The wax sections were eliminated in xylol:ethanol (1:1, v/v) solution for 5 min and rehydrated in several decreasing concentrations of ethanol, including 100, 95, 85 and 70%, for 1 min each concentration. The sample slides were stained in 3% safranin for 24 h and washed with a series of increasing ethanol concentrations of 70, 85 and 95%, for 5 s each concentration. Next, the sample slides were dipped into 1% fast green dye solution for 3 s each, treated twice with 100% of ethanol for 5 s each, and transferred into xylol:ethanol (1:1, v/v) solution for 5 min. All slides were mounted with synthetic resin and observed under a LSM510 laser scanning system (Carl Zeiss, Wetzlar, Germany).
Samples for scanning electron microscopy were prepared as above. Sample segments were gently dipped into xylol at 45 °C for 4 h to clean the wax. The samples were treated with xylol:ethanol (1:1, v/v) solution and 100% ethanol for 1 h and then put in tert-butyl alcohol. Finally, dried segments were metalized with gold for scanning electron microscope observation.
Protein extraction
Protein extraction was performed according to the previous study [28]. Approximately 0.5-g exocarp samples were kept in N2 and pulverized. The fine powder was suspended with pre-cooled lysis buffer, containing 8 M urea, 2 mM EDTA, 10 mM DTT and 1% Protease Inhibitor Cocktail, on ice overnight. The precipitate was removed at 15,000 g by centrifugation at 4 °C for 15 min. The final supernatants were redissolved in buffer containing 8 M urea, and 100 mM triethylammonium bicarbonate (TEAB) and quantified by a 2-D Quant kit (GE Healthcare, Beijing, China).
Digestion and tandem mass tag (TMT) labeling
The soluble samples were dissolved in 10 mM dithiothreitol buffer for 1 h at room temperature. The sample solution was alkylated with 20 mM iodoacetamide for 30 min at 25 °C in darkness. Then, the samples were redissolved in 100 μL of TEAB (100 mM). For the first digestion, trypsin (Sequencing Grade Modified Trypsin, V5113, Promega) was added at 1:50 (trypsin:protein) overnight and 1:100 (trypsin:protein) for a second digestion for 4 h. Approximately 200 μg of protein for each sample was treated for further experiments. The digestion was carried out at 25 °C.
Then, samples were desalted by a Strata X 33 μm C18 SPE column (8B-S100-AAK, Phenomenex, Torrance, CA, USA) and reconstituted using a 6-plex TMT kit (Thermo-Scientific, Rockford, USA). The supernatants were collected by centrifugation at 10,000×g for 15 min. Finally, the supernatants were subsequently labeled randomly with different TMT agents (Thermo-Scientific). Specifically, the amount of TMT-label solution needed to label 100 μg protein sample was reconstituted in 24 μL of acetonitrile. The mixture was incubated at room temperature for 2 h. At last, samples were pooled and dried b for further analysis.
Fractionation and LC–MS/MS analysis
The peptide samples were separated into various fractions by reverse-phase HPLC using an Agilent 300Extend C18 column (5 μm particles, 4.6 mm ID, 250 mm length, Santa Clara, CA, USA). The samples were detected at wavelength 250 nm. Then, the peptides were combined into 18 fractions and dried.
Fractionated samples were dissolved in 0.1% formic acid (FA) solution and directly loaded onto a Acclaim PepMap 100 reversed-phase pre-column (25 cm length, 2 μm, Thermo, Shanghai, China). The gradient comprised increases in solution B, containing 0.1% FA in 98% ACN, from 4 to 22% in 26 min, from 22 to 35% in 8 min, climbing to 80% in 3 min and maintained at 80% for 3 min, with a constant flow rate of 320 nL/min using an EASY-nLC 1000 UPLC system (Thermo, Shanghai, China). The peptide samples were subjected to an Nano-Spray Ionization source followed by MS/MS in a Q Exactive™ (Thermo, Shanghai, China) coupled to the online UPLC [29]. The MS proteomics data have been set to the ProteomeXchange Consortium by PRIDE partner repository program under identifier PXD008871.
Validation of DAPs by parallel reaction monitoring (PRM)
The PRM method was used to confirm the changes in DAPs identified in the LC–MS–TMT analysis. In this experiment, three independent samples were identified by an acquired MS/MS spectrum. Three biological replicates were used. The tryptic peptides were dissolved in solution (0.1% FA), directly loaded onto a reversed-phase analytical column (150 mm length, 75 μm, Thermo, Shanghai, China). The gradient of solution B containing 0.1% FA and 98% CAN, which comprised an increasing contents of 6 to 25% during 38 min, from 25 to 38% in 14 min, climbing to 80% in 4 min, and maintained at 80% for 4 min, with a constant flow rate of 400 nL/min.
Database search
The MS/MS data were processed using MaxQuant with integrated Andromeda search engine v.1.5.2.8. MS/MS data were searched against the papaya genome data (http://phytozome.jgi.doe.gov/pz/portal.html) concatenated with a reverse decoy database. Trypsin/P was used as cleavage enzyme allowing up to two missing cleavages. For precursor ions, the mass error was set to 10 ppm, and for fragment ions the mass error was set to 0.02 Da. For protein quantification, TMT 6-plex was selected in MaxQuant. For peptide and protein identification, the false discovery rate (FDR) threshold was adjusted to < 1% and the peptide ion score was set to ≥20.
Annotation methods
Gene ontology (GO) annotation of our proteome was derived from the UniProt-GOA database (www. http://www.ebi.ac.uk/GOA/). Firstly, all identified protein IDs were converted to UniProt IDs and mapped to GO IDs. Then, InterProScan soft was used to annotate the proteins that were not annotated by UniProt-GOA database based on protein sequence alignment method.
Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/) was used to annotate protein pathways. Firstly, KEGG online service tools KAAS (http://www.genome.jp/tools/kaas/) was used to annotate the KEGG description of proteins. Then, the annotation results were mapped to the KEGG pathway database using online tool KEGG Mapper (http://www.kegg.jp/kegg/mapper.html).
A subcellular localization predication software, WOLFPSORT, was used to predict subcellular localization (https://wolfpsort.hgc.jp/).
Functional enrichment analysis
Within the DAPs, a Fisher’s test was used to analyze the GO and KEGG functional enrichments. Correction hypothesis was carried out using the FDR control method. GO and KEGG categories with a corrected P-value < 0.05 were considered significant. K-means cluster was analyzed using the MeV software. In order to meet the requirements of the hierarchical clustering method [30].
Fatty acid content determination
Fatty acid methyl ester was prepared according to the previous study [31] with several minor alterations. Three biological replicates were used. A 40-mg fruit sample was put into a 15-mL tube. Then, 2 ml of mixture of ethyl ether and n-hexane (1:1, v/v), 2 ml of 2-mol/l KOH in methanol solution and 2 ml of methanol were added to the glass tube. The mixture solution was kept in a water bath at 50 °C for 30 min with shaking. When the solution was cool, 2 mL of deionized water was added to the tube. The up layer was collected and loaded to GC–MS. A constant flow rate of 0.8 mL/min was used as the carrier gas. The temperature of oven was set initially at 170 °C for 30 s, then raised to 230 °C at a rate of 5 °C/min and kept at 230 °C for 13 min. The MS was applied in an electron mode at 70 eV with a scan range of 30–450 m/z.
Results
Micromorphology and physiology changes of papaya fruit during ripening
Two groups of papaya fruit (five per group) at the time points of 0 and 8 d after harvest were collected and analyzed. Papaya peel during ripening exhibited morphological changes in the epidermal and sub-epidermal cells. Fruit at 0 d had a glossy surface and at 8 d had an irregular surface (Fig. 1a–d). On 0 day, peel was comprised of epidermal cells with bulliform shape (Fig. 1e). By day 8, the upper layers showed horizontal elongation with an associated decrease in cell height (Fig. 1f). The firmness of fruit was rapidly down-regulated during ripening. Respiration rate of fruit was much higher in 8 d than that in 0 d. The papaya had a significantly higher TSS and lower TA during fruit ripening (Fig. 1g–i).

Micromorphology and physiology changes of papaya fruit during ripening process. Pictures of papaya fruit under time points D0 (a) and D8 (b) after harvest. Light microscopy observation of surface tissues collected from the fruit under time points D0 (c) and D8 (d) after harvest. Scanning electron microcopy observation of surface tissues collected from the fruit under time points D0 (e) and D8 (f) after harvest. The physiological features, including firmness (g), respiration rate (h), total soluble solids (i) and titratable acidity (j), of papaya fruit under time points after harvest
Quality control and quantitative proteome analysis
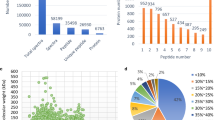
The changes in the proteome of papaya fruit at two different ripening stages were quantified. The correlation coefficients of two stages × three replicates indicated a good repeatability of MS data (Fig. 2a). The mass errors of the identified peptides were measured – most mass errors were < 0.02 Da, and their distribution was near zero (Fig. 2b). The lengths of all identified peptides were within 8–16 Aa (Fig. 2c). To get more detailed information on the identified and quantified proteins, GO, KEGG, domain and subcellular localization annotations of all proteins were showed in Additional file 1.

Quantitative proteome analysis and QC validation of MS data. Protein were extracted in three biological replicates for each fruit group. Proteins were trypsin digested and analyzed by HPLC-MS/MS. (a) Pearson’s correlation of protein quantitation. (b) Mass delta of all identified peptides. (c) Length distribution of all identified peptides
Identification of DAPs during fruit ripening
A total of 960 DAPs, including 425 up- and 535 down-regulated proteins, were identified (Additional files 2 and 3). Among the DAPs, the top five up-regulated proteins were a triphosphate hydrolase superfamily protein (9.3 fold), followed by an inhibitor of trypsin and hageman factor-like protein (8.4 fold), a groES-like zinc-binding dehydrogenase family protein (7.7 fold), an 1-aminocyclopropane-1-carboxylate oxidase (7.6 fold) and a glutamine aminotransferase (7.6 fold). The top five down-regulated proteins were three chlorophyll a–b binding proteins (34.5, 21.2 and 14.7 fold, respectively), a PsaB protein (16.7 fold) and a Photosystem I 11 K family protein (15.6 fold) (Additional file 3). Subcellular locations of the DAPs were predicted (Additional file 4). For the induced proteins, a total of 11 different groups were identified, such as chloroplast- (146 proteins), cytosol- (122), nucleus- (58), mitochondria- (29) and plasma membrane-localized proteins (28 proteins) (Additional file 2). For reduced proteins, 14 components were identified, including chloroplast- (193 proteins), cytosol- (167), nucleus- (58), extracellular- (25) and plasma membrane-localized protein (25) (Additional file 2).
Enrichment analysis of DAPs during fruit ripening
In total, 332 DAPs were assigned to at least one GO term. For up-regulated proteins, the highly enriched ‘Molecular Function’ GO terms were ‘alpha-amino acid biosynthetic process’, ‘cellular amino acid biosynthetic process’, ‘carboxylic acid biosynthetic process’, ‘pigment metabolic process’, ‘cellular lipid metabolic process’, ‘isoprenoid metabolic process’, ‘organic acid biosynthetic process’ and ‘small molecule biosynthetic process’; and the significantly enriched ‘Biological Process’ GO terms were ‘Photosystem I’, ‘Photosystem II’, ‘thylakoid membrane’, ‘extrinsic component of membrane’, ‘photosynthetic membrane’ and ‘thylakoid part’ (Fig. 3a). For down-regulated proteins, the highly enriched ‘Biological Process’ GO terms were related to ‘Photosystem I’, ‘Photosystem II’ and ‘thylakoid membrane’; within the ‘Molecular Function’, the most significantly enriched terms were ‘calcium ion binding’, ‘endopeptidase inhibitor activity’ and ‘endopeptidase regulator activity’; and the most enriched terms in the ‘Cellular Component’ were ‘cell wall modification’, ‘photosynthetic electron transport chain’ and ‘photosynthesis, light reaction’ (Fig. 3b).

GO enrichment analysis of DAPs. (a) Distribution of the up-regulated proteins with GO annotation. Different color blocks represent different terms, including cellular component, molecular function, and biological process. Number of the up-regulated proteins in each second level term was showed in a pie chart. (b) Distribution of the down-regulated proteins with GO annotation. Different color blocks represent different terms, including cellular component, molecular function, and biological process. Number of the down-regulated proteins in each second level term was showed in a pie chart
The up-regulated proteins were mostly associated with ‘Biosynthesis of secondary metabolites’ and ‘Fatty acid biosynthesis’; and the down-regulated proteins were mostly involved in ‘Photosynthesis’ and ‘Glyoxylate and dicarboxylate metabolism’ (Additional file 5).
The up-regulated proteins mostly contained a ‘Isopenicillin N synthase-like’ domain and down-regulated proteins mostly contained a ‘Chlorophyll a/b binding protein’ domain (Additional file 6).
Involvement of pathogen defense-related proteins in the fruit ripening process
We identified a number of pathogen defense-related proteins: five pathogenesis-related (PR) proteins, four tubulin (TUB) family proteins, one suppressor of G2 allele of SKP1 (SUGT) protein, one plant–pathogen interacting (PTI) protein, four nucleolin (NCL) proteins, five heat shock proteins (HSPs), one glycerol kinase (glpK) protein, four calcium-Dependent protein kinase (CPK) proteins, three cyclic nucleotide gated channel (CNGF) proteins, five calcium-binding proteins (CMLs), two calmodulin (CALM) proteins and one brassinosteroid-insensitive 1-associated receptor kinase 1 (BAK1) (Additional file 7). Then, the subcellular locations of these pathogen defense-related proteins were predicted, mostly in chloroplast, cytosol and nucleus. Additionally, two CNGFs and one CML were predicted as plasma membrane located, one PR protein was extracellularly located and one HSP90 protein was located in vacuolar membrane (Fig. 4a).

Analysis of the pathogen defense-related proteins during the papaya fruit ripening process. (a) The subcellular locations of these pathogen defense-related proteins in papaya. (b) Relative expression levels of the proteins related to pathogen defense. Significant differences in expression level were indicated by “*”
Among these pathogen defense-related proteins, nine were quantified as DAPs during papaya fruit ripening and, interestingly, most were significantly down-regulated (Fig. 4b).
Involvement of fatty acid metabolism in the fruit ripening process
Three fatty acid related metabolism pathways were significantly changed during the ripening process of papaya fruit: ‘Fatty acid biosynthesis’, ‘Fatty acid metabolism’ and ‘Fatty acid degradation’ (Fig. 5a). For the ‘Fatty acid biosynthesis’ pathway, two down-regulated and 11 up-regulated proteins were identified (Fig. 5b); for the ‘Fatty acid metabolism’ pathway, four down-regulated and 13 up-regulated (Fig. 5c); and for the ‘Fatty acid degradation’ pathway, four down-regulated and five up-regulated (Fig. 5d). Enzymes involved in fatty acid metabolism are listed in Additional file 8.

Identification and expression analysis of the KEGG terms related to Fatty acid metabolism. (a) KEGG enrichment analysis of DAPs. Differential expression profiling of ‘fatty acid biosynthesis’-(b), ‘fatty acid metabolism’-(c), and ‘fatty acid degradation’-related genes (d) under the fruit ripening process. Red indicated the number of up-regulated genes and green indicated the number of down-regulated genes
Furthermore, the contents of several important fatty acids were determined during the fruit ripening process. In total, 16 kinds of fatty acids were identified and respectively quantified at 0 and 8 d (Fig. 6). Notably, four fatty acids – methyl-decanoate, methyl laurate, methyl palmitoleate and methyl α-octadecatrienoic acid – were significantly up-regulated at 8 d compared with 0 d; and four fatty acids – methyl stearate, methyl oleate, methyl erueate and methyl linoleate – were significantly down-regulated. Contents of the remaining eight fatty acids did not significantly change.

The changes in the contents of various fatty acids during the papaya fruit ripening process. Significant differences in contents were indicated by “*”
Involvement of cell wall-related proteins in the fruit ripening process
A number of enzymes, including PG, pectinesterase (PE), α-galactosidase (α-GAL), β-GAL, β-xylosidase, PL, expansin (EXP), xyloglucan-endotransglycosylase (XET) and endoglucanase (EG), are known to be involved in cell wall metabolism [32]. In our study, three PGs, six PEs, two α-GALs, one β-GAL, two EXPs, two XETs and two EGs were identified as DAPs. The expression patterns of the cell wall metabolism-related proteins were thus analyzed during the ripening process: all PGs, α-GALs and most PEs (except for Unigene0023139) were significantly down-regulated; and all β-GAL, EXPs, EGs and one EXT (Unigene0032975) were significantly up-regulated (Additional file 9).
Verification of the changes in fruit ripening-related proteins using PRM
Because fruit ripening is extremely complex, it is difficult to identify a single sensitive biomarker. Instead, the identification of a panel of candidate enzymes that reflect the process of fruit ripening would offer a better understanding of this complex process. To validate the differential expression of several key enzymes involved in papaya fruit ripening, PRM was applied. In total, 16 key proteins, including five fatty acid metabolism-related, two pathogen defense-related, two ethylene biosynthesis-related and seven cell wall biosynthesis-related proteins, were selected for PRM verification (Additional file 10). The relative abundances of several key proteins from different sample groups are presented in Figs. 7 and 8. The trend of these DAPs determined by PRM was consistent with our TMT-label quantification results.

Verification of the changes in fatty acid- and pathogen defense-related proteins using PRM. Five representative proteins, including three proteins involved in fatty acid metabolism (a), two pathogen defense-related proteins (b), were randomly selected for PRM verification. For each protein, the abundances of two peptides were determined

Verification of the changes in ethylene biosynthesis- and cell wall-related proteins using PRM. Nine representative proteins, including two proteins involved in ethylene biosynthesis (a), and two cell wall-related proteins (b), were randomly selected for PRM verification. For each protein, the abundances of two peptides were determined
Discussion
Fruit ripening involves a process of several modifications, including starch–sugar conversion, cell wall degradation, flavor substance and pigment biosynthesis, and metabolic rearrangement [32,33,34]. As climacteric fruit, softening of papaya is an important feature that determines its marketing value [35]. Increasing numbers of studies have focused on molecular and biochemical mechanisms underlying the regulation of climacteric fruit ripening. In our study, comprehensive analysis was carried out to identify the DAPs involved in the ripening process.
Several proteomic analyses of papaya fruit have been performed using the 2DE method. For example, proteomic analysis of PMeV-infected papaya identified a total of 486 reproducible spots, among which calreticulin and the proteasome subunits 20S and RPT5a were upregulated during infection [36]. A differential analysis of the proteomes of climacteric and non-climacteric papaya fruit revealed 27 proteins with high differences in levels during ripening [37]. Comparative proteomic analysis of somatic embryo identified 76 spots, suggesting a role of polyethylene glycol in somatic embryo development in papaya [26]. We identified a total of 3220 proteins and 2818 proteins were quantified, which is more than in previously published proteomes of papaya fruit [25, 37]. Embracing comprehensive information allowed us to explore new proteins/genes that were potentially involved, directly and indirectly, in the ripening process.
A sharp increase in ethylene production is considered a marked feature of climacteric fruits at the initial stage of ripening [38]. In higher plants, the important roles of two enzymes, ACC synthase and ACC oxidase, in ethylene biosynthesis have been well-studied [39]. In papaya, two different ACO genes were identified as participating in papaya fruit ripening [40]. In the up-regulated protein list, the most remarkable were two ACC oxidases, up-regulated during the ripening process by 7.6 and 1.8 fold, respectively (Additional file 1). A similar result was also reported in a 2DE-DIGE analysis [37]. Our data confirmed the critical role of ethylene in papaya fruit ripening.
The ripening of fleshy fruit is associated with changes in susceptibility to pathogen infection and resistance to pathogen infection significantly decreases during ripening [41]. Pathogen-related proteins (PRs) play a role in multiple functions, particular in defense against pathogens [42]. In papaya fruit, eight pathogen-related factors were significantly reduced and only one significantly induced (Fig. 4b), indicating that the disease resistance of papaya fruit might decrease during ripening.
The metabolic shifts throughout ripening cause alterations in primary and secondary metabolites, such as amino acids, fatty acids, sugars, polyphenols and cinnamic acids [43]. Our KEGG enrichment analysis revealed that various metabolic pathways were significantly changed during ripening, suggesting a close relationship between metabolism and fruit ripening of papaya. Flavonoids, the ubiquitous fruit secondary metabolites, play important roles in coloring, abiotic stress defense and other biological functions [44]. In date palm, proteins involved in anthocyanin biosynthesis were up-regulated particularly from the onset of ripening and during ripening [45]. Similarly, during the ripening of papaya, several flavonoid biosynthesis-related proteins were up-regulated: cinnamate-4-hydroxylase, coumaroyl 3-hydroxylase and flavanone-3-hydroxylase (Fig. 5 and Additional file 1). Increases in flavonoids may provide raw materials for pigment biosynthesis, accelerating the color variation of papaya fruit [46].
Interestingly, three fatty acid metabolic pathways were highlighted by our KEGG enrichment analysis, and a number of fatty acid metabolism-related enzymes were also identified as DAPs (Fig. 5). Studies on the changes in fatty acid composition during fruit development and ripening have been performed for various fruit plants. For example, the levels of ω-6 and ω-3 fatty acids significantly changed during mango fruit development and ripening [47]. Ripening raises the content of polyunsaturated fatty acids in tomato fruit [48]. In our study, assessment and profiling assessed the variation of fatty acids during the ripening process of papaya fruit (Fig. 6). Among 16 quantified fatty acids, eight showed significant changes during the ripening process (Fig. 6). In tomato fruit, ripening increases the contents of unsaturated fatty acids [48]. In papaya, two unsaturated fatty acids, methyl palmitoleate and methyl α-octadecatrienoic acid, were significantly up-regulated during ripening. Increases in unsaturated fatty acids might be associated with papaya fruit volatile formation [49].
Conclusions
In summary, a total of 3220 proteins were detected, 2828 of these were quantified. Among the DAPs, most pathogen-related proteins were down-regulated during the ripening process. Moreover, flavonoid and fatty acid metabolisms were greatly changed during ripening; and unsaturated fatty acids may be associated with papaya fruit volatile formation. Our data may give an intrinsic explanation of the variations in metabolism and offer important information about how to expend the shelf life of postharvest papaya fruit.
Availability of data and materials
The datasets generated and analysed during the current study are available in the ProteomeXchange Consortium by PRIDE partner repository program under identifier PXD008871.
Abbreviations
- 1-MCP:
-
1-methylcyclopropene
- 2-DE:
-
two-dimensional gel electrophoresis
- ABA:
-
abscisic acid
- ACO:
-
ACC oxidase
- BAK1:
-
brassinosteroid-insensitive 1-associated receptor kinase 1
- CALM:
-
calmodulin
- CML:
-
calcium-binding proteins
- CNGF:
-
cyclic nucleotide gated channel
- CPK:
-
calcium-Dependent protein kinase
- DAP:
-
differential accumulated protein
- EG:
-
endoglucanase
- EXP:
-
expansin
- FA:
-
formic acid
- FAA:
-
formalin acetic acid
- glpK:
-
glycerol kinase
- GO:
-
Gene ontology
- HPLC:
-
high-performance liquid chromatography
- HSP:
-
heat shock protein
- JA:
-
jasmonic acid
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LC–MS/MS:
-
liquid chromatography-mass spectrometer
- NCL:
-
nucleolin
- PE:
-
pectinesterase
- PR:
-
pathogenesis-related
- PRM:
-
Parallel Reaction Monitoring
- PTI:
-
plant–pathogen interacting
- SUGT:
-
suppressor of G2 allele of SKP1
- TEAB:
-
triethylammonium bicarbonate
- TUB:
-
tubulin
- XET:
-
xyloglucan-endotransglycosylase
- α-GAL:
-
α-galactosidase
- β-GAL:
-
β-galactosidase
References
Giovannoni JJ. Genetic regulation of fruit development and ripening. The Plant cell, vol. 16 Suppl; 2004. p. S170–80.
Wang WQ, Møller IM, Song SQ: Proteomic analysis of embryonic axis of Pisum sativum seeds during germination and identification of proteins associated with loss of desiccation tolerance. J Proteomics Journal of proteomics 2012, 77(24):68–86.
Brummell DA. Cell wall disassembly in ripening fruit. Funct Plant Biol. 2006;33(2):103–19.
Carpita NC, Gibeaut DM. Structural models of primary cell walls in flowering plants: consistency of molecular structure with the physical properties of the walls during growth. The Plant journal : for cell and molecular biology, vol. 3; 1993. p. 1):1–30.
Brummell DA, Harpster MH. Cell wall metabolism in fruit softening and quality and its manipulation in transgenic plants. Plant Mol Biol. 2001;47(1–2):311–40.
Yong W, Link B, O'Malley R, Tewari J, Hunter CT, Lu CA, et al. Genomics of plant cell wall biogenesis. Planta. 2005;221(6):747–51.
Dumville JC, Fry SC. Solubilisation of tomato fruit pectins by ascorbate: a possible non-enzymic mechanism of fruit softening. Planta. 2003;217(6):951–61.
Fry SC, Dumville JC, Miller JG. Fingerprinting of polysaccharides attacked by hydroxyl radicals in vitro and in the cell walls of ripening pear fruit. The Biochemical journal. 2001;357(Pt 3:729–37.
Jha SN, Jaiswal P, Narsaiah K, Kaur PP, Singh AK, Kumar R. Textural properties of mango cultivars during ripening. J Food Sci Technol. 2013;50(6):1047–57.
Xi W, Zheng H, Zhang Q, Li W. Profiling taste and aroma compound metabolism during apricot fruit development and ripening. Int J Mol Sci. 2016;17(7):pii: E998.
Yao BN, Tano K, Konan HK, Bedie GK, Oule MK, Koffi-Nevry R, et al. The role of hydrolases in the loss of firmness and of the changes in sugar content during the post-harvest maturation of Carica papaya L. var solo 8. J Food Sci Technol. 2014;51(11):3309–16.
Baldwin EA, Scott JW, Shewmaker CK, Schuch W. Flavor trivia and tomato aroma: biochemistry and possible mechanisms for control of important aroma components. Hortscience. 2000;35(6):1013–22.
Villa-Rodríguez JA, Molina-Corral FJ, Ayala-Zavala JF, Olivas GI, González-Aguilar GA. Effect of maturity stage on the content of fatty acids and antioxidant activity of ‘Hass’ avocado. Food Res Int. 2011;44(5):1231–7.
Lalel HJD, Singh Z, Tan SC. Aroma volatiles production during fruit ripening of ‘Kensington Pride’ mango. Postharvest Biology & Technology. 2003;27(3):323–36.
Liu K, Wang J, Li H, Zhong J, Feng S, Pan Y, et al. Identification, expression and IAA-amide synthetase activity analysis of Gretchen Hagen 3 in papaya fruit (Carica papaya L.) during postharvest process. Front Plant Sci. 2016;7:1555.
Zou Y, Zhang L, Rao S, Zhu X, Ye L, Chen W, et al. The relationship between the expression of ethylene-related genes and papaya fruit ripening disorder caused by chilling injury. PLoS One. 2014;9(12):e116002.
Paull RE. Postharvest atemoya fruit splitting during ripening. Postharvest Biology & Technology. 1996;8(4):329–34.
Liu K, Yuan C, Li H, Lin W, Yang Y, Shen C, et al. Genome-wide identification and characterization of auxin response factor (ARF) family genes related to flower and fruit development in papaya (Carica papaya L.). BMC Genomics. 2015;16:901.
Fabi JP, Seymour GB, Graham NS, Broadley MR, May ST, Lajolo FM et al: Analysis of ripening-related gene expression in papaya using an Arabidopsis-based microarray. BMC Plant Biol 2012, 12:242–242.
Othman R, Nuraziyan A. Fruit-specific expression of papaya subtilase gene. J Plant Physiol. 2010;167(2):131–7.
Shiga TM, Fabi JP, do Nascimento JR, Petkowicz CL, Vriesmann LC, Lajolo FM, et al. Changes in cell wall composition associated to the softening of ripening papaya: evidence of extensive solubilization of large molecular mass galactouronides. J Agric Food Chem. 2009;57(15):7064–71.
do Prado SBR, Melfi PR, Castro-Alves VC, Broetto SG, Araújo ES, do Nascimento JRO, et al. Physiological degradation of pectin in papaya cell walls: release of long chains galacturonans derived from insoluble fractions during postharvest fruit ripening. Front Plant Sci. 2016;7:1120.
Fabi JP, Broetto SG, da Silva SL, Zhong S, Lajolo FM, do Nascimento JR. Analysis of papaya cell wall-related genes during fruit ripening indicates a central role of polygalacturonases during pulp softening. PLoS One. 2014;9(8):e105685.
Fu CC, Han YC, Qi XY, Shan W, Chen JY, Lu WJ, et al. Papaya CpERF9 acts as a transcriptional repressor of cell-wall-modifying genes CpPME1/2 and CpPG5 involved in fruit ripening. Plant Cell Rep. 2016;35(11):2341–52.
Huerta-Ocampo JA, Osuna-Castro JA, Lino-Lopez GJ, Barrera-Pacheco A, Mendoza-Hernandez G, De Leon-Rodriguez A, et al. Proteomic analysis of differentially accumulated proteins during ripening and in response to 1-MCP in papaya fruit. J Proteome. 2012;75(7):2160–9.
Vale EdM, Heringer AS, Barroso T, Ferreira ATdS, da Costa MN, Perales JEA et al: Comparative proteomic analysis of somatic embryo maturation in Carica papaya L. Proteome Sci 2014, 12:37–37.
Soares EA, Werth EG, Madronero LJ, Ventura JA, Rodrigues SP, Hicks LM, et al. Label-free quantitative proteomic analysis of pre-flowering PMeV-infected Carica papaya L. J Proteome. 2017;151:275–83.
Hao J, Guo H, Shi X, Wang Y, Wan Q, Song YB, et al. Comparative proteomic analyses of two Taxus species (Taxus × media and Taxus mairei) reveals variations in the metabolisms associated with paclitaxel and other metabolites. Plant & cell physiology. 2017;58(11):1878–90.
Xu D, Yuan H, Tong Y, Zhao L, Qiu L, Guo W, et al. Comparative proteomic analysis of the graft unions in hickory (Carya cathayensis) provides insights into response mechanisms to grafting process. Front Plant Sci. 2017;8:676.
Yu C, Guo H, Zhang Y, Song Y, Pi E, Yu C, et al. Identification of potential genes that contributed to the variation in the taxoid contents between two Taxus species (Taxus × media and Taxus mairei). Tree Physiol. 2017;37(12):1659–71.
He Z, Zhu H, Li W, Zeng M, Wu S, Chen S, et al. Chemical components of cold pressed kernel oils from different Torreya grandis cultivars. Food Chem. 2016;209:196–202.
Brownleader MD, Jackson P, Mobasheri A, Pantelides AT, Sumar S, Trevan M, et al. Molecular aspects of cell wall modifications during fruit ripening. Crit Rev Food Sci Nutr. 1999;39(2):149–64.
Xiao YY, Kuang JF, Qi XN, Ye YJ, Wu ZX, Chen JY, et al. A comprehensive investigation of starch degradation process and identification of a transcriptional activator MabHLH6 during banana fruit ripening. Plant Biotechnol J. 2017.
Huo W, Li B, Kuang J, He P, Xu Z, Wang J. Functional characterization of the steroid reductase genes GmDET2a and GmDET2b form Glycine max. Int J Mol Sci. 2018;19(3).
Paull RE, Chen NJ. Postharvest variation in cell wall-degrading enzymes of papaya (Carica papaya L.) during fruit ripening. Plant Physiol. 1983;72(2):382–5.
Rodrigues SP, Ventura JA, Aguilar C, Nakayasu ES, Almeida IC, Fernandes PM, et al. Proteomic analysis of papaya (Carica papaya L.) displaying typical sticky disease symptoms. Proteomics. 2011;11(13):2592–602.
Nogueira SB, Labate CA, Gozzo FC, Pilau EJ, Lajolo FM, Oliveira do Nascimento JR. Proteomic analysis of papaya fruit ripening using 2DE-DIGE. J Proteome. 2012;75(4):1428–39.
Liu M, Pirrello J, Chervin C, Roustan JP, Bouzayen M. Ethylene control of fruit ripening: revisiting the complex network of transcriptional regulation. Plant Physiol. 2015;169(4):2380–90.
López-Gómez R, Cabrera-Ponce JL, Saucedo-Arias LJ, Carreto-Montoya L, Villanueva-Arce R, Díaz-Perez JC, et al. Ripening in papaya fruit is altered by ACC oxidase cosuppression. Transgenic Res. 2009;18(1):89–97.
Chen YT, Lee YR, Yang CY, Wang YT, Yang SF, Shaw JF. A novel papaya ACC oxidase gene (CP-ACO2) associated with late stage fruit ripening and leaf senescence. Plant Sci. 2003;164(4):531–40.
Cantu D, Blanco-Ulate B, Yang L, Labavitch JM, Bennett AB, Powell AL. Ripening-regulated susceptibility of tomato fruit to Botrytis cinerea requires NOR but not RIN or ethylene. Plant Physiol. 2009;150(3):1434–49.
Jia X, Zeng H, Wang W, Zhang F, Yin H. Chitosan oligosaccharide induces resistance to Pseudomonas syringae pv. Tomato DC3000 in Arabidopsis thaliana by activating both salicylic acid- and jasmonic acid-mediated pathways. Molecular plant-microbe interactions. MPMI. 2018;31(12):1271–9.
Liang Z, Sang M, Fan P, Wu B, Wang L, Duan W, et al. Changes of polyphenols, sugars, and organic acid in 5 Vitis genotypes during berry ripening. J Food Sci. 2011;76(9):C1231–8.
Broun P. Transcriptional control of flavonoid biosynthesis: a complex network of conserved regulators involved in multiple aspects of differentiation in Arabidopsis. Curr Opin Plant Biol. 2005;8(3):272–9.
Marondedze C, Gehring C, Thomas L. Dynamic changes in the date palm fruit proteome during development and ripening. Horticulture research. 2014;1:14039.
Mou W, Li D, Luo Z, Mao L, Ying T. Transcriptomic analysis reveals possible influences of ABA on secondary metabolism of pigments, flavonoids and antioxidants in tomato fruit during ripening. PLoS One. 2015;10(6):e0129598.
Deshpande AB, Chidley HG, Oak PS, Pujari KH, Giri AP, Gupta VS. Data on changes in the fatty acid composition during fruit development and ripening of three mango cultivars (alphonso, Pairi and Kent) varying in lactone content. Data in Brief. 2016;9:480–91.
Saini RK, Zamany AJ, Keum YS: Ripening improves the content of carotenoid, alpha-tocopherol, and polyunsaturated fatty acids in tomato (Solanum lycopersicum L.) fruits. 3 Biotech 2017, 7(1):43.
Wang JJ, Liu HR, Gao J, Huang YJ, Zhang B, Chen KS. Two omega-3 FADs are associated with peach fruit volatile formation. Int J Mol Sci. 2016;17(4):464.
Acknowledgements
We are grateful to Dr. Xiaolin Zheng (Zhejiang Gongshang University), Dr. Zhiping Deng (Zhejiang Academy of Agricultural Sciences) and Dr. Jiang Tian (South China Agricultural University) for technical support. The authors declare that they have no competing interests.
Funding
This work was funded by the Science and Technology Program of Guangdong, China (2015A020208018 and 2017A040403069); Natural Science Foundation of Guangdong Province, China (2016A030307016 and 2018A0303130161); Science and Technology Project of Zhanjiang City, China (2016A03016) and College Student Climbing Program of Guangdong Province, China (pdjh2019b0299). There is on role of the funding bodies in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
All authors have read and approved the final version. JW, LX and KL conceived and designed the study. BJ, LX, TZ, CY and JW collected and taken care of the plant samples. BJ, LX, SO, WM, MY, and HG performed the experiments. CY analyzed the data. CS and KL wrote the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This project uses plant materials and does not utilize transgenic technology. The seedlings of papaya tree were purchased by the Yixin horticulture company (Zhanjiang, China), who provided permission to use the seedlings for our scientific research. Collection of plant material complied with the Convention on the Trade in Endangered Species of Wild Fauna and Flora: https://www.cites.org/.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. The detail information of the identified proteins. (PDF 316 kb)
Additional file 2:
Figure S1. Variations in protein abundances during the papaya fruit ripening. (XLSX 747 kb)
Additional file 3:
Table S2. The detail information of the DAPs during fruit ripening. (XLSX 118 kb)
Additional file 4:
Table S3. The scores for the subcellular location of each protein. (XLSX 141 kb)
Additional file 5:
Figure S2. KEGG enrichment analysis of the DAPs during the ripening process of papaya fruits. (PDF 175 kb)
Additional file 6:
Figure S3. Protein domain enrichment analysis of the DAPs during the ripening process of papaya fruits. (PDF 308 kb)
Additional file 7:
Table S4. The detail information of the pathogen defense-related proteins. (XLSX 17 kb)
Additional file 8:
Table S5. The information of the fatty acid metabolism-related proteins. (XLSX 14 kb)
Additional file 9:
Table S6. The information of the proteins involved in the cell-related process. (XLSX 10 kb)
Additional file 10:
Table S7. The PRM results of 16 key proteins. (XLSX 13 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jiang, B., Ou, S., Xu, L. et al. Comparative proteomic analysis provides novel insights into the regulation mechanism underlying papaya (Carica papaya L.) exocarp during fruit ripening process. BMC Plant Biol 19, 238 (2019). https://doi.org/10.1186/s12870-019-1845-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1845-4