
Abstract
Background
Glioblastoma (GBM) is the most common malignant intracranial tumor with a low survival rate. However, only few drugs responsible for GBM therpies, hence new drug development for it is highly required. The natural product Cudraflavone B (CUB) has been reported to potentially kill a variety of tumor cells. Currently, its anit-cancer effect on GBM still remains unknown. Herein, we investigated whether CUB could affect the proliferation and apoptosis of GBM cells to show anti-GBM potential.
Results
CUB selectively inhibited cell viability and induced cell apoptosis by activating the endoplasmic reticulum stress (ER stress) related pathway, as well as harnessing the autophagy-related PI3K/mTOR/LC3B signaling pathway. Typical morphological changes of autophagy were also observed in CUB treated cells by microscope and scanning electron microscope (SEM) examination. 4-Phenylbutyric acid (4-PBA), an ER stress inhibitor, restored the CUB-caused alteration in signaling pathway and morphological change.
Conclusions
Our finding suggests that CUB impaired cell growth and induced cell apoptosis of glioblastoma through ER stress and autophagy-related signaling pathways, and it might be an attractive drug for treatment of GBM.
Similar content being viewed by others

Background
GBM is the most common adult brain tumor (55%) with a high malignancy degree [1]. The common therapeutic strategies of GBM include surgery, radiation therapy, and chemotherapy [2]. Even though tremendous trials have been made to treat GBM [3, 4], the prognosis of GMB patients is still poor. The median survival time of newly diagnosed patients is less than 15 months.
Recently, several natural product-derived compounds including flavonoids have drawn more attention as potential agents for adjuvant GBM targeted therapies [5, 6]. The preliminary experiments suggest that natural products could harness the GBM cells via inducing apoptosis, increasing reactive oxygen species (ROS), inhibiting metastasis and regulating unfolded protein response (UPR)[7,8,9]. Moreover, the natural products, Apigenin-7-O-β-d-(-6′′-p-coumaroyl)-glucopyranoside (APG), caused severe and persistent ER stress in tumor cells leading to cell death [10, 11].
Cudraflavone B (CUB) is a flavonoid compound found in a variety of plants (Fig. 1a), first extracted and purified from Cephalotaxus fortune [12]. It has been applied to anti-aging in clinical due to its distinct antioxidant activity [13]. Furthermore, studies have shown that CUB induced apoptosis in human oral cancer cells and melanoma cells in vivo [14, 15]. In addition, CUB lead to a blockage of cell cycle sub-G1 and exhibited an inhibition effect on Monoamine oxidase (MAO) activity in mice [16]. All these findings indicated that CUB may have the potential to treat GBM. However, the experimental evidence is still lacking, and the underlying mechanisms remain elusive.

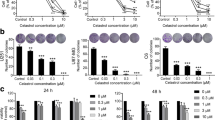
CUB increases apoptosis of GBM cells. a The structure of CUB. b The effects of CUB on cell viability of U87, U251 GBM cell lines and normal human astrocytes cells. c, d Clone formation assay of U87 c and U251 d cells treated with CUB. e Apoptosis measurements of GBM cells with CUB (10, 20 μM) incubation for 12(Additional file: Fig. S1a), 24 h. Data represents the mean ± SEM (n = 3). Statistical analysis was performed using one-way ANOVA. *P < 0.05, **P < 0.01, and ***P < 0.001
Based on these conditions, we engaged in the research for CUB in GBM. Here, we investigated whether CUB can affect the growth of GBM and explore the molecular mechanisms underlying its anti-GBM activity. We first tested the effect of CUB on the viability and proliferation in U87, U251 and normal human astrocyte cell lines. Next, transcriptome sequencing was used to identify the gene expression changes in CUB-treated cells. Finally, functional and mechanism experiments were conducted to further verify the molecular mechanisms underlying the anticancer effects of CUB in GBM.
Materials and methods
Cell lines and cell culture
Human GBM cell lines and normal human astrocytes(NHA), U87 and U251, were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China). Cells were cultured in high glucose Dulbecco's modified Eagle's medium (DMEM) (Hyclone, USA) supplied with 10% fetal bovine serum (FBS) (Gibco, USA) and 100 μM streptomycin, 100 U/mL penicillin. All cells were cultured at 37 ℃ with 5% CO2 and 95% humidity in a constant temperature incubator. CUB (CUB, C25H24O6, Relative molecular mass: 420.5, purity > 94%, yellow powder) was purchased from WuXi PharmaTech (WuXi, China), which was dissolved in dimethyl sulphoxide (DMSO) at 30 mM as stored concentration and was diluted in DMEM for usage.
Cell viability
Cell viability was detected using a cell-titer blue kit (Promega, USA). U87 and U251 cells (1 × 104 cells/well) were seeded in 96-well plates and cultured for 24 h, then cells were treated with different concentrations of CUB for 48 h. To detect cell viability, 20 μl cell-titer blue reagents were diluted in 100 μl DMEM medium. In reverse experiments, cells were pre-treated with 4-PBA, 3-MA or CQ for 12 h before CUB treatment. Cell viability was quantified using Odyssey CLx Infrared Imaging System.
Cloning formation assay
Cells were seeded in 6 well plates (1 × 103 cells/well) and exposed to 5 μM CUB for 14 days. To stain the colony, cells were washed three times by PBS, then fixed with 4% polyformaldehyde (PFA) for 30 min and subsequently stained with 1% crystal violet for 5 min. Colonies were quantified using Odyssey CLx Infrared Imaging System.
Apoptosis analysis
U87 and U251 cells were seeded in 6 well plates (4 × 105 cells per well) and cultured for 24 h to achieve 90–100% confluence. After reagents treatment, annexin V-propidium iodide (AV-PI) staining was conducted to assess apoptosis rate following the manufacturer’s instructions (Beyotime, Shanghai, China). Cell apoptosis was detected by flow cytometry (FC) (BD Biosciences, AccuriTM C6; San Jose, CA, USA) and analyzed using the Flowjo software (Tree Star; Ashland, OR, USA).
Western blot analysis
Cells were lysed in Radio Immuno Precipitation Assay (RIPA) buffer and the cell extraction was collected after centrifugation. Cell extraction was mixed with 5 × sodium dodecyl sulfate polyacrylamide gel electropheresis (SDS-PAGE) loading buffer (Beyotime, Shanghai, China), then boiled at 98 ℃ for 10 min. Proteins were separated by SDS–polyacrylamide gel electrophoresis then transferred to 0.22 μm Polyvinylidene Fluoride (PVDF) membranes (Millipore, Boston, MA, USA). Membranes were blocked for 2 h in Tris-buffered saline containing 5% non-fat dry milk (Biosharp, China), then cut it into several blots to hybridized with the indicated primary antibodies (1:1000 dilution) at 4 ℃ overnight. Horseradish peroxidase secondary antibody (1:2000) was incubated for approximately 2 h at room temperature then detected by the immobile western chemiluminescent horseradish peroxidase (HRP) substrate (Super ECL Detection, Yeasen, China) (ABclonal, China).
Real-time PCR
RNA was extracted after 24 h post-drug treatment. The total RNA of samples was extracted using EZ-press RNA Purification Kit (EZBioscience, USA). A 4 × Reverse Transcription Master Mix (EZBioscience) was used for reverse transcription reaction at 42 ℃ for 15 min, 95 ℃ for 30 S. The 2 × SYBR Green qPCR Master Mix (EZBioscience) was used to perform qPCR following this protocol: denaturation (5 min, 95 ℃), and 40 amplification cycles (10 s at 95 ℃, and 30 s at 60 ℃) by using the Strata Gene Mx3000p (Agilent Technologies, Inc., Santa Clara, CA). Each gene of interest was normalized to glyceraldehyde phosphate dehydrogenase (GAPDH) and the fold change was compared relative to the control sample. Each assay was performed in triplicate and experiments were repeated in at least three pooled cell samples.
SEM examination
Cells were fixed by 4% glutaraldehyde and postfixed in 1% OsO4 in 0.1 M cacodylate buffer for 2 h. After being stained with 1% Millipore-filtered uranyl acetate, the samples were then dehydrated in increased concentrations of ethanol, then infiltrated and embedded in epoxy resin (ZXBR, Spon 812). Electron photomicrographs of GBM cell ultra-structures were taken with a scanning electron microscope (JEM-1200EX II, JEOL; Tokyo, Japan).
Transferase-mediated deoxyuridine triphosphate-biotin nick end labeling (TUNEL) analysis
TUNEL staining was performed using a TUNEL apoptosis detection kit (Servicebio, Wuhan). After CUB treatment, cells were immersed in 4% PFA solution for 15 min then washed with PBS. Next, proteinase K solution (20 μg/ml) was added for 30 min they washed with PBS. A 50-μl Equilibration Buffer was added to each sample for 10 min. Then added 56 μl TdT-labeled reaction mixture (the mixture was compounded: Recombinant TdT enzyme:CF640-dUTP Labeling Mix: Equilibration Buffer = 1 µl:5 µl:50 µl (1:5:50). The cells were placed in a wet box for 60 min at 37 °C then washed with Phosphate Buffered Saline (PBS). Images were taken under fluorescence microscopy (Leica, Japan) and five different fields were randomly selected. Three independent assays were conducted. The cell numbers were calculated using ImageJ-5.0 software (Windows, 64-bit Java 1.8.0_112). The TUNEL-positive rate equals the number of TUNEL-positive cells divided by the total cell number × 100%.
Immunofluorescence (IF) staining
Immunofluorescence staining was performed in GBM cell lines growing in dishes of NEST® cell culture (China). According to a standard protocol, ice-cold 4% PFA served as fixative. Primary antibodies were rabbit polyclonal Abs against human activating transcription factor 4 (ATF4) and C/EBP-homologous protein (CHOP) (Abclonal). Nuclei were stained with DAPI (4,6-diamidino-2′-phenylindol, 5 µg/ml). Secondary Abs were goat anti-rabbit Alexa Fluor 488 IgG and goat anti-mouse Alexa Fluor 647 (Invitrogen, Camarillo, CA).
Transcriptome sequencing and expression analysis
U87 cells were treated with CUB then the total RNA was extracted as whole transcriptome libraries, and deep sequenced by Novogene Bioinformatics Technology Cooperation (Beijing, China). The protocol of differential expression analyses of genomics and Feature Counts of genomic features were described accordingly in previous study [17,18,19,20].
mRFP-GFP-LC3 transfection
The adenovirus probe mRFP-GFP-LC3 was used to examine autophagic flux. The transfected cells were incubated with 4% paraformaldehyde. Autophagic flux was observed under Nikon microscope (Tokyo, Japan).
Acridine orange (AO) staining
Cell apoptosis and lysosomal membrane permeability were evaluated by the AO staining assay (Sigma Aldrich, USA), when red fluorescence was emited in the acidic secondary lysosomes and diffuse green fluorescence in the cytoplasm [21]. Cells were incubated 5 μg/ml AO for 15 min, and observed under a Nikon microscope (Tokyo, Japan).
Statistical analysis
The data were analyzed using GraphPad Prism 7.0. All data presented as means ± SEM. Statistical analysis was conducted by one-way analysis of variance (ANOVA). The statistical significances labeled as *P < 0.05, **P < 0.01, ***P < 0.001.
Results
CUB inhibited cell growth and induced apoptosis of GBM cells.
To determine the effect of CUB (Fig. 1a) on GBM cells, two GBM cell lines U87 and U251 were treated with different concentrations of CUB (5, 10, 20, 40, 80 μM) for 24 h. The Cell Titer-Blue results showed that GBM cells viability decreased significantly after CUB treatment, compared with normal human astrocytes (Fig. 1b). It also suggests that CUB suppressed cell survival in a dose-dependent manner, while the major differences between tumor cells and normal cells were observed at 10 and 20 μM in 24 h. In addition, cloning formation assays confirmed that CUB inhibited GBM cell survival ((Fig. 1c, d). Furthermore, we validated the CUB’s effect on apoptosis of the GBM cells. GBM cells were treated with CUB as indicated, then stained with Annexin V and PI for further flow cytometry analysis. CUB-induced cell apoptosis was found in a dose-dependent and time-dependent manner in both U87 and U251 cells (Fig. 1e, Additional file 1: Fig. S1a).
CUB activated ER stress pathways in human GBM cells
Knowing that CUB could use to harness GBM cells, the underlying mechanism is still unknown. To address that, an RNA-seq analysis was employed. U87 cells were treated with 10 μM CUB for 24 h. Then the total RNA of treated and untreated cells was extracted for the transcriptome test. RNA-seq analysis revealed that the gene expression levels of ER stress and UPR pathways were significantly higher after CUB treatment (Fig. 2a, b). We next checked a group of ER stress-related genes. The expression level of phosphorylation of eukaryotic initiation factor-2α (eIF2α), CHOP and ER-stress sensor inositol-requiring enzyme 1(IRE1) were increased upon CUB treatment in a dose-dependent manner (Fig. 2c). To confirm this, we detected the ER stress-related protein level, CHOP and p-eIF2α. Indeed, it is very consistent with the result of real-time PCR (Fig. 2d).

CUB activates ER stress. a, b Gene Ontology (GO) pathways a and Kyoto Encyclopedia of Genes and Genomes (KEGG) b analyzed the transcriptome sequencing of U87 GBM cells after CUB treatment. U87 cells were treated with 10 μM of CUB. c Quantitative real-time PCR (qPCR) analysis of GBM cells treated with of CUB (5,10, 20 µM) for 24 h. d Immunoblotting of the ER stress markers, p-eIF2Α and CHOP, in GBM cells (Notion: here are cropped blots, original cropped images and replicates are presented in Additional file 1: Fig. S2). e Fluorescence images of ATF4 in GBM cells exposed to CUB (10 μM) or control for 24 h. Data were mean ± SEM from three independent experiments. The statistical analysis was performed using one-way ANOVA, and the significance labeled as * P < 0.05, ** P < 0.01 and *** P < 0.001
In addition, immune fluorescence assays demonstrated that CUB cloud active ATF4 expression in U87 cells (Fig. 2e). In conclusion, our findings indicated that CUB triggers severe ER stress via PERK-eIF2α-ATF4 pathways.
CUB induced autophagy and inhibited PI3K/mTOR/LC3B pathway
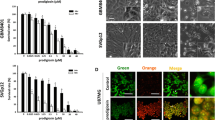
ER stress often lead to a dramatic change of cell morphologies [22]. Indeed, after treatment with CUB for 24 h, GBM cells exhibited swelling, cytoplasmic vacuolization and cellular tentacles retraction (Fig. 3a). Among these, the cytoplasmic vacuoles accumulation is one of the typical characteristics of cell autophagy [23, 24]. To verify this, we used scanning SEM to characterize vacuole formation in U87 and U251 cells treated with 20 μM CUB for 12, 24 h. We observed the typical autophagy phenotype, the double-membrane structure of autophagic vacuoles and large cytoplasmic vacuoles on electron micrographs (Fig. 3c, Additional file 1: Fig. S1b). To further confirm whether CUB could cause autophagy in GBM cells, we performed mRFP-GFP-LC3 transfection assay and AO staining. According to the confocal fluorescence imaging, CUB treatment increased the LC3 II level in U87 and U251 cells, as indicated by increased GFP + mRFP + /GFP + mRFP- ratio and raised AO fluorescence intensity (Additional file 1: Fig. S3a, b), as well as increased ratio of LC3-II/ LC3I (Fig. 3b). Additionally, p-mTOR, p-p70 and p-Akt were down-regulated while mTOR, p70 and Akt were unchanged (Fig. 3b). Taken together, these data suggested that CUB activated the autophagy in GBM cells with classical morphological changes and harnessed PI3K/Akt/mTOR/LC3B signaling pathway.

CUB induces GBM cell autophagy. a Morphological images of GBM cells. b Western blot analysis detected markers of the Akt/mTOR/pS70k pathway and LC3B-I, LC3-II in U251 cells treated with CUB at indicated concentration for 24 h (10 µg of the cell lysates was loaded)(Cropped blots, original cropped images and replicates are presented in Additional file 1: Fig. S5 and S6). c SEM images of U251, U87 cells treated with CUB (10, 20 µM) or DMSO for 24 h. Boxes highlight autophagic vacuoles and ER stress bubbles. Scale bars were marked in the images
3-Methyladenine(3-MA), PI3K inhibitor, was selected to inhibit the autophagy. To verify if ER stress-induced autophagy cause cell death, we performed the pre-treatment of 3-MA for 4 h to detect the autophagic flux and AO staining. We observed that 3-MA suppressed autophagic flux and prevented CUB-induced cell apoptosis (Additional file 1: Fig. S3a, b). To further verify CUB could induce autophagy-dependent cell death, we added another autophagy inhibitor chloroquine (CQ). CQ pretreatment reversed the inhibition of PI3K/Akt/mTOR pathway by CUB with increased phosphorylation levels of mTOR and P70 (Additional file 1: Fig. S4a) in U251 and U87 cells, and protected U87 cells from the cytotoxic effects of CUB (Additional file 1: Fig. S4b). Taken together, our results indicate that CUB promotes autophagy to induce cell death through inhibition of PI3K/AKT/mTOR signaling.
4-PBA could rescue the CUB triggered ER stress and autophagy-related phenotype
To better understand the roles of ER stress signaling in CUB-mediated ER stress and autophagy, we investigated whether abolish the ER stress-related pathways could reverse the CUB-mediated phenotype. The GBM cells were pre-treated with 4-PBA (an ER stress inhibitor) for 12 h before CUB treatment. Then, we checked the ER stress- and autophagy-related protein levels. As expected, the ER stress inhibitor 4-PBA counteracted the expression change of p-eIF2α, CHOP and Akt/mTOR/LC3B caused by CUB (Fig. 4a, b). Correspondingly, ER stress vacuoles reduced distinctly in 4-PBA pretreated cells (Fig. 4c). In addition, 4-PBA increased the viability of GBM cells, which was decreased by CUB treatment (Fig. 4d). To further verify the effect of 4-PBA on cell apoptosis, we conducted TUNEL assay. In line with cell viability results, 4-PBA counteracted CUB in apoptosis (Additional file 1: Fig. S7a). These observations confirmed that CUB-induced apoptosis via activating the ER stress related pathways.

The ER stress inhibitor 4-PBA counteracts CUB. a, b Western blot detected the protein levels of p-eIF2Α, CHOP Akt/mTOR/pS70k pathway, LC3B, and β-actin in GBM cells pretreated with 4-PBA (2 mM) for 12 h before exposure to CUB (20 µM)(Cropped blots, original cropped images and replicates are presented in Additional file 1: Fig. S8 and. S9). c Morphological images of U87 cells treated with CUB (10 µM), 4-PBA (2 mM), and CUB (10 µM) with 4-PBA (2 mM) together, respectively (Cropped blots). d Cell viability of GBM cells CUB (10 µM), 4-PBA (2 mM) and CUB (10 µM) with 4-PBA (2 mM) together. The scale bars were marked in the image. All data were the mean ± SEM of values from experiments performed in triplicate. * P < 0.05, ** P < 0.01 and *** P < 0.001 compared to control
Discussion
Natural products produced from the plants are important sources of screening leading compounds in cancer treatment. To date, more than half of the FDA-approved anti-tumor drugs come from natural products and their derivatives [25]. In this study, we revealed that natural flavonoid CUB suppressed cell growth and promoted cell apoptosis in GBM. Moreover, we investigated the underlying molecular mechanisms, and found that CUB stimulates ER stress-dependent autophagy in human GBM cells via the PERK/eIF2 pathway. Inhibition of CUB-induced ER stress can prevent autophagy and restored cell vitality. Together, these data suggested that CUB is a potential drug candidate for GBM therapy.
Temozolomide (TMZ) was used to treat GBM for over a decade, but its treatment benefits are limited due to resistance [26]. CUB has been reported to show anti-proliferative activity in B16 melanoma cells, human gastric carcinoma cells and human oral cancer cells with effective lowest concentration at the 12uM in previous experiments. Our studies showed that CUB induced cell apoptosis in both U87 and U251 GBM cells by ER stress-induced autophagy, here we showed that its IC50 was 10uM, which indicated it is promising for in vivo experiments and clinical studies.
In our study, CUB induced ~ 56% cell death at the dose of 20uM after 24 h in U87 cells, while ~ 40% cell death in the case of U251(Fig. 1a). We found U87 displayed earlier response to CUB- induced ER stress as evidenced by the earlier appearance of intracellular vacuoles after 12 h treatment of CUB (Additional file 1: Fig. S1c). Previous research reported that although U87 and U251 cells showed high ratios of PI3K-AKT signaling pathway activation, U251 cells are more resistant to hypoxia and low glucose condition [27]. The glycolysis rate of cells plays a vital role in mitochondrial respiration function to determine the relative contribution of autophagy, which is consistent with the lower autophagy level that was observed for U251 cells compared with U87 cells in another study (Additional file 1: Fig. S1c). This may reflect that U251 cells has a slowly autophagy response to ER stress induced by CUB.
Although CUB shows promising anticancer-activity in multiple cancers types via inducing apoptosis, our knowledge of its underlying molecular mechanisms is very limited, e.g. regulating the tumour necrosis factor α (TNFα) and nuclear fractor-κB (NF-κB) expression, affecting the anti-inflammatory response [28], and activating the MAPK signaling pathway to promote apoptosis [13]. Here, we observed ER stress was induced by CUB treatment in GBM cells. Based on transcriptome analysis results, we demonstrated that the CUB-induced GBM cell apoptosis is dependent upon persistent ER stress that activates the PERK/ATF4/CHOP pathway. It is very well in line with that persistent ER stress causes tumor apoptosis, provides prospects for tumor treatment strategies [22, 29]. In addition, we also observed cell cycle inhibition upon CUB. This is consistent with a previous study that in response to PERK-ATF4-CHOP UPR reaction, eIF2α is activated to inhibit cyclin-D1 expression, causing cell cycle G1 blocking [30].
Furthermore, our results support that drug-induced persistent ER stress resulted in cell death via autophagy [31]. CUB treated cells showed the classic morphological and molecular characteristics of autophagy, such as autolysosomes formation and the membrane-binding LC-II. [32]. Mechanically, the result highlighted CUB-induced ER stress-activated autophagy through Akt/mTOR/RPS6KB1 pathway. In line with this, the ER stress inhibitor increased the activity of Akt/mTOR/RPS6KB1 signaling, leading to less autophagy flux. These results highlighted this pathway as a potential mediator of CUB-induced autophagy in GBM cells. Considering the dysregulated and reprogrammed metabolic environment in cancer cells, it needs deep understanding for the PERK/ATF4/CHOP in crosstalk between ER stress and apoptosis.
However, inhibition of ER stress does not fully abrogate cell death. This may be because excess CUB impairs cell viability through activation of NF-κB (Additional file 1: Fig. S7b). Additionally, it has shown impressive anti-inflammatory and neuro-protective effects on macrophages. In GBM, tumor-associated macrophages comprise half of tumor mass and involving in tumor progression [33]. We believe that it will be worth further exploration through its promotion effect on macrophages and resistance to tumors, if it can cross the blood–brain barrier. Currently, the concentrations of CUB we used are close to those used in other anti-tumor reports. However, we did not conduct in vivo experiment to assess its permeability to the blood–brain barrier and suitable administration concentration. We insight it will be further explored in the next studies.
Availability of data and materials
The datasets generated during and analyzed during the current study are not publicly available due to we propose to continue our study of this drug in GBM therapy but are available from the corresponding author on reasonable request.
Abbreviations
- GBM:
-
Glioblastoma
- CUB:
-
Cudraflavone B
- APG:
-
Apigenin-7-O-β-d-(-6″-p-coumaroyl)-glucopyranoside
- UPR:
-
Unfolded protein response
- ROS:
-
Reactive oxygen species
- 4-PBA:
-
4-Phenylbutyric acid
- MAO:
-
Monoamine oxidase
- NHA:
-
Normal human astrocytes
- FBS:
-
Fetal bovine serum
- DMSO:
-
Dimethyl sulphoxide.
- FC:
-
Flow cytometry
- RIPA:
-
Radio immuno precipitation assay
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electropheresis
- PVDF:
-
Polyvinylidene fluoride
- HRP:
-
Horseradish peroxidase
- GAPDH:
-
Glyceraldehyde phosphate dehydrogenase
- TUNEL:
-
Transferase-mediated deoxyuridine triphosphate-biotin nick end labeling
- PBS:
-
Phosphate buffered saline
- IF:
-
Immunofluorescence
- CHOP:
-
C/EBP-homologous protein
- IRE1:
-
ER-stress sensor inositol-requiring enzyme 1
- eIF2α:
-
Eukaryotic initiation factor-2α
- KEGG:
-
Kyotoe ncyclopedia of genes and genomes
- GO:
-
Gene ontology
- qPCR:
-
Quantitative real-time PCR
- TMZ:
-
Temozolomide
- TNFα:
-
Tumour necrosis factor α
- CQ:
-
Chloroquine
References
Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR, Gottardo N, Gutmann DH, Hargrave D, Holland EC, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol. 2019;16(8):509–20.
Brinkman TM, Krasin MJ, Liu W, Armstrong GT, Ojha RP, Sadighi ZS, Gupta P, Kimberg C, Srivastava D, Merchant TE, et al. Long-term neurocognitive functioning and social attainment in adult survivors of pediatric CNS tumors: results from the St Jude lifetime cohort study. J Clin Oncol. 2016;34(12):1358–67.
Le Rhun E, Preusser M, Roth P, Reardon DA, van den Bent M, Wen P, Reifenberger G, Weller M. Molecular targeted therapy of glioblastoma. Cancer Treat Rev. 2019;80:101896.
Moreau A, Febvey O, Mognetti T, Frappaz D, Kryza D. Contribution of different positron emission tomography tracers in glioma management: focus on glioblastoma. Front Oncol. 2019;9:1134.
Park MN, Song HS, Kim M, Lee MJ, Cho W, Lee HJ, Hwang CH, Kim S, Hwang Y, Kang B, et al. Review of natural product-derived compounds as potent antiglioblastoma drugs. Biomed Res Int. 2017. https://doi.org/10.1155/2017/8139848.
Santos BL, Oliveira MN, Coelho PL, Pitanga BP, da Silva AB, Adelita T, Silva VD, Costa Mde F, El-Bacha RS, Tardy M, et al. Flavonoids suppress human glioblastoma cell growth by inhibiting cell metabolism, migration, and by regulating extracellular matrix proteins and metalloproteinases expression. Chem Biol Interact. 2015;242:123–38.
Su TR, Tsai FJ, Lin JJ, Huang HH, Chiu CC, Su JH, Yang YT, Chen JY, Wong BS, Wu YJ. Induction of apoptosis by 11-dehydrosinulariolide via mitochondrial dysregulation and ER stress pathways in human melanoma cells. Mar Drugs. 2012;10(8):1883–98.
Ma J, Yang YR, Chen W, Chen MH, Wang H, Wang XD, Sun LL, Wang FZ, Wang DC. Fluoxetine synergizes with temozolomide to induce the CHOP-dependent endoplasmic reticulum stress-related apoptosis pathway in glioma cells. Oncol Rep. 2016;36(2):676–84.
Xipell E, Gonzalez-Huarriz M, Martinez de Irujo JJ, Garcia-Garzon A, Lang FF, Jiang H, Fueyo J, Gomez-Manzano C, Alonso MM. Salinomycin induced ROS results in abortive autophagy and leads to regulated necrosis in glioblastoma. Oncotarget. 2016;7(21):30626–41.
Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40(3):141–8.
Feng Y, Lu Y, Liu D, Zhang W, Liu J, Tang H, Zhu Y. Apigenin-7-O-beta-d-(-6’’-p-coumaroyl)-glucopyranoside pretreatment attenuates myocardial ischemia/reperfusion injury via activating AMPK signaling. Life Sci. 2018;203:246–54.
Fujimoto T, Hano Y, Nomura T, Uzawa J. Components of root bark of Cudrania Tricuspidata 2. structures of two new Isoprenylated flavones Cudraflavones A and B. Planta Med. 1984;50(2):161–3.
Hosek J, Bartos M, Chudik S, Dall’Acqua S, Innocenti G, Kartal M, Kokoska L, Kollar P, Kutil Z, Landa P, et al. Natural compound Cudraflavone B shows promising anti-inflammatory properties in vitro. J Nat Prod. 2011;74(4):614–9.
Arung ET, Yoshikawa K, Shimizu K, Kondo R. Isoprenoid-substituted flavonoids from wood of Artocarpus heterophyllus on B16 melanoma cells: Cytotoxicity and structural criteria. Fitoterapia. 2010;81(2):120–3.
Tano T, Okamoto M, Kan S, Nakashiro K, Shimodaira S, Yamashita N, Kawakami Y, Hamakawa H. Growth inhibition and apoptosis by an active component of OK-432, a streptococcal agent, via Toll-like receptor 4 in human head and neck cancer cell lines. Oral Oncol. 2012;48(8):678–85.
Lee DS, Ko W, Kim DC, Kim YC, Jeong GS. Cudarflavone B provides Neuroprotection against glutamate-induced mouse Hippocampal HT22 cell damage through the Nrf2 and PI3K/Akt signaling pathways. Molecules. 2014;19(8):10818–31.
Dong C, Li X, Yang J, Yuan D, Zhou Y, Zhang Y, Shi G, Zhang R, Liu J, Fu P, et al. PPFIBP1 induces glioma cell migration and invasion through FAK/Src/JNK signaling pathway. Cell Death Dis. 2021;12(9):827.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51.
Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb). 2021;2(3):100141.
Ni Y, Gu WW, Liu ZH, Zhu YM, Rong JG, Kent TA, Li M, Qiao SG, An JZ, Zhang HL. RIP1K contributes to neuronal and astrocytic cell death in ischemic stroke via activating autophagic-lysosomal pathway. Neuroscience. 2018;371:60–74.
Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529(7586):326–35.
Fulda S, Kogel D. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene. 2015;34(40):5105–13.
Wu R, Murali R, Kabe Y, French SW, Chiang YM, Liu S, Sher L, Wang CC, Louie S, Tsukamoto H. Baicalein targets GTPase-mediated autophagy to eliminate liver tumor-initiating stem cell-like cells resistant to mTORC1 inhibition. Hepatology. 2018;68(5):1726–40.
Wang XB, Liu W, Yang L, Guo QL, Kong LY. Investigation on the substitution effects of the flavonoids as potent anticancer agents: a structure-activity relationships study. Med Chem Res. 2012;21(8):1833–49.
Chen X, Zhang M, Gan H, Wang H, Lee JH, Fang D, Kitange GJ, He L, Hu Z, Parney IF, et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat Commun. 2018;9(1):2949.
Li H, Lei B, Xiang W, Wang H, Feng W, Liu Y, Qi S. Differences in protein expression between the U251 and U87 cell lines. Turk Neurosurg. 2017;27(6):894–903.
Lee HJ, Auh QS, Lee YM, Kang SK, Chang SW, Lee DS, Kim YC, Kim EC. Growth inhibition and apoptosis-inducing effects of Cudraflavone B in human oral cancer cells via MAPK, NF-kappa B, and SIRT1 signaling pathway. Planta Med. 2013;79(14):1298–306.
Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–97.
Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci USA. 2000;97(23):12625–30.
Green DR, Levine B. To be or not to be? how selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75.
Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155(6):1216–9.
Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, Rasmussen R, Dwivedi B, Seby S, Wolf SA, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 2017;77(9):2266–78.
Acknowledgements
We are grateful to J.Y. and Y.Z. for insightful comments on this study, and RB.Z. for their SEM experiment technical assistance.
Funding
This work was supported by the Young Scientists Fund of the National Natural Science Foundation of China (81903028), Innovative and Entrepreneurial Talent Program of Jiangsu for Guohua Shi team, the China Postdoctoral Science Foundation (2020M671599), Projects of International Cooperation of Jiangsu Province (No. BZ2020004). Suzhou municipal key clinical disciplines cultivate program (SZFCXK202142), Suzhou Medical Innovation Applied Research Project (SKYD2022090).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Jinlin Pan, Jiao Yang, Caihua Dong and Rongchuan Zhao, Yanxiang Liu, Yuanshuai Zhou. The bioinformatic analysis was provided by Yuanshuai Zhou. The SEM assays was performed by Roubing Zhang. The first draft of the manuscript was written by Jinlin Pan. The draft was critically revised by Nafees Ahmad, Yanxiang Liu, Yuanshaui Zhou and Minxuan Sun. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Authors state no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Fig. S1.
CUB induced autophagy through ER stress-dependent pathways. (a)Flow cytometry analysis results of U87, U251 cells at 12h treatment of CUB (10μM, 20μM). (b) Representative SEM images of U87, U251 cells occurred autophagy induced by CUB (10μM, 20μM). (c)Morphological images of GBM cells at 6h of CUB treatment. n = 3 per group. Means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. Fig. S2. CUB activates ER stress, blots origin images and replicates of Fig2d. Fig. S3. CUB activates autophagy flux in GBM cells. (a) Confocal images and the ratio of GFP-LC3/mRFP-LC3 at 12h. CUB (10μM), 3-MA(10mM). (b) Confocal images of AO staining at 12h. Fig. S4. CUB induced cell death through autophagy-dependent pathways. (a) Western blotting analysis of U87, U251 cells at 12h treatment of CUB (0μM, 20μM) or 3-MA(1mM) and CQ(30μM) pretreatment followed treatment of CUB(20μM). (b) Autophagy inhibitors suppressed U87 cell proliferation detected by the Cell-title Blue assay. Cells were incubated with 3-MA(1mM) or CQ (30μM) 12 h and then cultured in CUB(20μM) medium for the indicated times. Means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Fig. S5. CUB induces GBM cell autophagy, blots origin images and replicates of Fig. 3b (U87). Fig. S6. CUB induces GBM cell autophagy, blots origin images and replicates of Fig. 3b (U251). Fig. S7. CUB promoted apoptosis and NF-κB activation in U87 cells. (a) Images of TUNEL staining Green: TUNEL-positive cells, Blue: DAPI. (b) Western Blot analysis of NF-κB pathway. All data are expressed as the mean ± SEM of values from experiments performed in triplicate. * P < 0.05, ** P < 0.01 and *** P < 0.001 compared to control (Cropped blots). Fig. S8. The ER stress inhibitor 4-PBA counteracts CUB, blots origin images and replicates of Fig. 4a. Fig. S9. The ER stress inhibitor 4-PBA counteracts CUB, blots origin images and replicates of Fig. 4a. Fig. S10. CUB promoted apoptosis and NF-κB activation in U87 cells. blots origin images and replicates of Fig. S1b. Fig. S11. Part images of differential contrast blot
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pan, J., Zhao, R., Dong, C. et al. Cudraflavone B induces human glioblastoma cells apoptosis via ER stress-induced autophagy. BMC Neurosci 24, 10 (2023). https://doi.org/10.1186/s12868-023-00778-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12868-023-00778-4