
Abstract
Background
Quorum sensing (QS) is a cell density-based intercellular communication system that controls virulence gene expression and biofilm formation. In Pseudomonas aeruginosa (P. aeruginosa), the LasR system sits at the top of the QS hierarchy and coordinates the expression of a series of important traits. However, the role of lasR in phage infection remains unclear. This study aims to investigate the role of lasR QS in phage infection.
Methods
The P. aeruginosa phage was isolated from sewage, and its biological characteristics and whole genome were analyzed. The adsorption receptor was identified via a phage adsorption assay. Following lasR gene knockout, the adsorption rate and bactericidal activity of phage were analyzed. Finally, real-time quantitative polymerase chain reaction (RT-qPCR) was conducted to explore how lasR promoting phage infection.
Results
The lytic phage vB_Pae_PLY was isolated and lipopolysaccharide (LPS) was identified as its adsorption receptor. The adsorption rate and bactericidal activity of vB_Pae_PLY were reduced after lasR knockout. RT-qPCR results showed that the expression of galU, a key gene involved in LPS synthesis, was down-regulated, and several genes related to type IV pili (T4P) were also down-regulated in the lasR mutant PaΔlasR.
Conclusions
The study showed that QS lasR may promote phage vB_Pae_PLY infection by involving in the synthesis of LPS and T4P. This study provides an example of QS in promoting phage infection and deepens the understanding of phage-bacteria interactions.
Similar content being viewed by others

Background
Pseudomonas aeruginosa (P. aeruginosa) is an opportunistic pathogen that uses quorum sensing (QS) signaling molecules to regulate the expression of virulence genes and biofilm [1]. P. aeruginosa has a particularly complex QS signaling network, which primarily consists of four interconnected systems (LasI/R, RhlI/R, PQS, and IQS) [2]. In the Las and Rhl systems, the lasI and rhll genes are responsible for the synthesis of N -(3-oxo-dodecanoyl)- L -homoserine lactone (3- oxo -C12-HSL) and N -(butanoyl)- L -homoserine lactone (C4-HSL), respectively. Then, 3- oxo -C12-HSL and C4-HSL interact with LasR, respectively, to form LasR- 3- oxo -C12-HSL and RhlR- C4-HSL complex to regulate target genes [3]. The PQS system enables the biosynthesis of QS signal molecules 2-heptyl-4-hydroxyquinoline (HHQ) and 2-heptyl-3-hydroxy-4(1 H)-quinolone (PQS, Pseudomonas quinolone signal) [4]. IQS is the fourth and recently discovered QS system, which synthesizes the signaling molecule 2-(2-hydroxyphenyl)-thiazole-4-carbaldehyde (IQS) [1]. Among all the QS systems, the Las tops the signal network. The LasR- 3- oxo -C12-HSL complex positively regulates the expression of receptor and synthase genes in the downstream QS system, thus establishing a regulatory feedforward loop [5]. QS directly or indirectly regulates more than 10% of its genome and about 20% of its bacterial proteome [6, 7], and controls the expression of a series of important traits in P. aeruginosa [8, 9].
Phages are viruses that specifically infect bacteria and are abundant in environments associated with their host bacteria. Phages bind to the surfaces of bacterial cells by recognizing adsorption receptors [10]. Several factors relating to the host bacteria affect phage infection, including the surface structure, immune system, growth state, genetic variation, and quorum sensing (QS) [11,12,13]. Currently, several studies have reported the role of QS in phage infection of host bacteria. QS can regulate phage defense. P. aeruginosa used QS to activate the expression of its CRISPR-Cas immune defense system, thereby protecting the bacteria from phage infection [14, 15]. QS also reduced phage infection by down-regulating phage receptors [16, 17]. However, the study by Xuan et al. found that lasI QS promoted phage infection in P. aeruginosa [18]. In summary, the role of P. aeruginosa QS in phage-bacteria interactions is obviously diverse and complex. In the future, more studies are needed to reveal the role of P. aeruginosa QS in phage infection.
This research aims to elucidate the role and mechanism of lasR QS in phage infection. The study shows that the lasR QS may promote phage infection via the biosynthesis of lipopolysaccharide (LPS) and type IV pili(T4P), key components of bacterial surface required for phage adsorption and infection. This finding will help to fully understand the mechanism of phage infection under QS,and guide the application of phage therapy.
Materials and methods
Strains, plasmids, and growth conditions
Comprehensive details of the strains and plasmids used in this study are shown in Table S1 in the supplementary materials. All primers used for polymerase chain reaction (PCR) in this study are listed in Table S2. P. aeruginosa was cultured in blood plate or Luria-Bertani (LB) broth medium at 37℃. Carbenicillin (Car, 300 µg/mL) and gentamicin (Gen, 200 µg/mL) (Wenzhou Kangtai Biological Technology Co., Zhejiang, China) were added as needed.
Isolation of the phage
Referring to the methodology outlined in a previous study [19], the phage vB_Pae_PLY was isolated from the sewage of the First Affiliated Hospital of Wenzhou Medical University using PAO1 as the host strain and enriched to reach a titer of 1 × 1010 plaque-forming units (PFU)/mL. The purified phage lysate was stored in SM buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, and 8 mM MgSO4) at 4℃ for later use. Then, the double-layer agar plate method was used to determine phage titer. Briefly, 100 µL of 109 colony-forming units (CFU)/mL bacterial host culture was mixed with 100 µL of ten-fold serially diluted phage lysate and incubated for 10 min at room temperature. Subsequently, 8 mL of melted 0.4% soft agar maintained at 45 °C was added to the phage–bacteria mixture. After mixing, the contents of the tube were immediately poured over a pre-prepared confluent monolayer of 1.5% agar. After incubation at 37℃ for 8–12 h, the number of PFUs was calculated. Serially diluted phage suspensions were prepared in triplicate for each dilution factor. The titer of the phage stock (PFU/mL) was calculated as follows:
Titer (PFU/mL) = Number of PFUs × 10 (Volume conversion factor) × Dilution factor.
Analysis of biological characteristics of the phage
Host range determination
Spot assays were performed to determine the host range of the vB_Pae_PLY phage against an additional 40 strains of P. aeruginosa clinical isolates. The presence of a clear zone and lysis plaque was recorded as the tested strain being susceptible to the phage [20].
One-step growth curve
The one-step phage growth was performed as previously described but with few modifications [21]. The phage vB_Pae_PLY was mixed with the host P. aeruginosa PAO1 with a multiplicity of infection (MOI) of 0.01. After adsorption at 37℃ for 10 min, the mixture was centrifuged at 12,000 × g for 2 min to remove the unadsorbed phages in the supernatant. The pellet was washed twice with LB broth medium, resuspended in 25 mL of fresh LB broth medium, and incubated with shaking at 37°C, 180 rpm. The samples were then collected at 0, 10, 20, 30, 40, 60, 90, 120 and 240 min, respectively, and the phage titer was determined via the double-layer agar method. The burst size was calculated as the ratio of the final phage titer to the number of initial bacterial cells infected.
Transmission Electron Microscopy (TEM)
TEM imaging was performed following a previously established method to observe the morphology of phages [19].
Whole genome analysis of phage
Genomic structural analysis and functional annotation
The genomic DNA of vB_Pae_PLY was extracted using λ phage genomic DNA extraction kit (column type) (Beijing Abigen Biotechnology Co., Ltd., Beijing, China) according to the manufacturer’s instructions. Genome sequencing and analyses were performed at Shanghai Personal Biotechnology Co., Ltd. The genome was annotated using Prokka and eventually corrected manually by using BLASTP to search for similar proteins in the NCBI non-redundant database. The genomic architecture of vB_Pae_PLY was analyzed using Proksee (https://proksee.ca/) and the genome map was visualized [22]. Virulence factors and antibiotic resistance-encoding genes were searched using the Virulence Factor Database (VFDB, http://www.mgc.ac.cn/VFs/main.htm) and the Comprehensive Antibiotic Resistance Database (CARD, https://card.mcmaster.ca/analyze/rgi) webservers, respectively. In addition, phage genome-based life cycle identification was performed using the PhageAI online platform (https://phage.ai/) [19]. The complete genome sequence of vB_Pae_PLY was deposited in the GenBank database under accession number OR689712.
Phylogenetic tree analysis
ViPTree (https://www.genome.jp/viptree/) was used to generate viral proteomic tree and classify viruses based on genome-wide similarity. A total of 349 genome sequences of all Pseudomonadota phages of the family Autophagoviridae were selected for the phylogenetic tree analysis by iTOL (https://itol.embl.de/).
Identification of the phage adsorption receptor
Periodate and proteinase K treatments
Bacterial cell surface LPS and outer membrane proteins were destroyed using periodate and proteinase K, respectively [23, 24]. The PAO1 strain was cultured overnight in LB broth medium at 37 °C to an OD600 of 1 (approximately 1 ~ 3 × 109 CFU/mL). The bacterial culture was then mixed with 100 mM sodium periodate (NaIO4) and incubated at 25 °C in the dark for 2 h. Similarly, a 0.2 mg/mL proteinase K solution (QIAGEN, Germany) was added to the bacterial culture, and the mixture was incubated at 37 °C for 3 h. Following both treatments, the mixture was centrifuged at 12,000 × g for 1 min. The cells were washed and re-suspended in 1 mL LB broth medium. A control without NaIO4 or proteinase K was included.
The phage adsorption rate was then measured: 10 µL of phage suspension (1 × 108 PFU/mL) was added to 1 mL each of the above bacterial suspensions respectively and incubated at 37 °C with shaking at 180 rpm for 10 min to allow sufficient adsorption of phages. After centrifugation at 12,000 × g for 1 min, the phage titer of in the supernatant was measured using the double-layer agar plate method as described above. As a control, the phage titer in the LB broth medium alone was set to 100%. The phage adsorption rate was calculated as follows: Phage adsorption rate (%) = [(Control phage titer - Residual phage titer in supernatant)/ Control phage titer] ×100%.
LPS adsorption assay
LPS was isolated from P. aeruginosa PAO1 using the LPS Extraction Kit. The LPS adsorption assay was performed with minor modifications, as previously described [18]. Briefly, 50 µL of the extracted LPS was added to 850 µL of LB broth medium, and mixed with 100 µL of phage solution (105 PFU/mL) at 37 °C for 10 min to allow adsorption. For the control sample, 50 µL of phosphate-buffered saline (PBS) was added to the LB broth medium and mixed with the phage. The phage titer in the supernatant and the phage adsorption rate were determined as described above.
Analysis of the effect of lasR on phage infection
Construction of the lasR knockout mutant (PaΔlasR)
The gene knockout of lasR was performed using a previously described method from PAO1 [25]. The plasmids and primers used for the gene knockout are listed in Table S1 and S2, respectively. The lasR mutant strain was screened by colony PCR and the target fragment was sequenced (Beijing Luhe Huada Gene Technology Co.,Ltd, China). Finally, transcriptome sequencing technology was used to verify the success of the lasR gene knockout.
Spot assay
To evaluate phage sensitivity, 3 µL aliquots of 10-fold serial dilutions (105–107 PFU) of each phage were spotted on a bacterial lawn on double-layer agar plates [26]. After incubation at 37 °C for 12 h, plaque formation was observed.
Adsorption rate assay
The phage adsorption assay was conducted following a previous methodology with minor modifications [27]. 100 µL of 107 PFU/mL phage solutions were mixed with 900 µL of PAO1 and PaΔlasR bacterial cultures (1 × 108 CFU/mL) separately at an MOI of 0.01, and incubated at 37 °C for 10 min to allow sufficient phage adsorption. The titer of free phage in the supernatant was determined and phage adsorption rates were calculated as described above.
Phage-killing experiment
To further compare the effect of the lasR gene on phage infection efficiency, time-killing experiments were performed to detect the number of viable bacteria during the phage-killing process with minor modifications based on previous method [19]. Firstly, the PAO1 and PaΔlasR strains were cultured overnight in fresh LB broth medium, and the bacterial suspensions were adjusted to 0.5 McFarland. Then, 30 µL of bacterial suspensions were added to 3 mL of LB broth medium, followed by the addition of 30 µL of phage (1 × 109 PFU/mL). Further, the mixture was incubated at 37 °C with shaking at 180 rpm for 12 h. Following this, 20 µL of the culture was collected at 4, 8, and 12 h intervals, respectively, for viable colony counts.
Real-Time quantitative PCR (RT-qPCR)
RT-qPCR was performed referring to a previous study [28]. Individual colonies of freshly cultured PAO1 and PaΔlasR strains were grown to the logarithmic phase in the LB broth medium. Then, bacterial RNA was extracted using RNAiso Plus. The total cDNA was synthesized using the PrimeScript™ RT Reagent Kit, and Real-time PCR was performed following the manufacturer’s instructions (Takara Biomedical Technology Co., Ltd.). The levels of mRNA of the target genes were normalized against rpsL. Using PAO1 as the control strain, the relative expression was calculated by the 2−ΔΔCt method. All primers used in this study are listed in Table S2.
Statistical analyses
All the data were expressed as mean ± standard deviation of at least three independent experiments. Statistical significance was determined using an independent two-tailed t-test. The P-values of < 0.05, < 0.01, and < 0.001 were denoted by *, **, and ***, respectively, whereas insignificance was denoted by ns. The GraphPad Prism 8.0 software was used for the statistical analysis.
Results
Morphology, host range, and one-step growth curve of vB_Pae_PLY
Phage plaques with clear morphology and distinct boundaries were obtained on PAO1 (Fig. 1a). TEM observations of the viral particles revealed that vB_Pae_PLY is a short-tailed phage, which had a polyhedral capsid and an almost invisible tail; thus, it was classified to the Autographiviridae family according to the International Committee on Taxonomy of Viruses (ICTV) [29]. The head diameter and tail length of vB_Pae_PLY were approximately 55 and 10 nm, respectively (Fig. 1b). The phage sensitivity analysis of the 40 strains of P. aeruginosa showed that vB_Pae_PLY was able to lyse 24 of 40 (60%) clinical isolates to form a clear plaque region on a double-layer agar plate (Fig. 2).

(a) Plaque formation on a double-layer agar plate of vB_Pae_PLY; (b) Morphology of vB_Pae_PLY under TEM.

Lytic activity of vB_Pae_PLY against 40 strains of P. aeruginosa. +, phage formed a clear zone or plaque; −, phage formed no clear zone nor plaque
A one-step growth curve was used to determine the burst size and latent period of viruses. From the one-step growth curve of the phage vB_Pae_PLY (Fig. 3), the latent period, defined as the time between infection and subsequent release of phage virions, was about 40 min, the outbreak period was about 200 min, and the burst size was more than 853 PFU per infected cell.

The one-step growth curve showed a latency period of 40 min and high burst size (up to 853 PFU/infected cell)
Genomic signature of vB_Pae_PLY
To better understand the phage vB_Pae_PLY, its genomic DNA was extracted and sequenced. The complete nucleotide sequence of the genome of vB_Pae_PLY has been deposited in the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) under accession number OR689712. As shown in Fig. 4, vB_Pae_PLY has a dsDNA genome with 43,757 bp and a G + C content of 62.12%. The genome was predicted to contain 64 coding DNA sequences (CDS), which were classified into six functional categories, including “tail protein”, “connector”, “head and packaging”, “lysis”, “DNA/RNA and nucleotide metabolism”, and “hypothetical proteins”. Three CDS encode “lysis” proteins (Rz-like spanin by CDS 4, endolysin by CDS 5, and holin by CDS 6). Among the 64 CDS, 34 encode hypothetical proteins that share similarities with other P. aeruginosa phage and have no clear function in the replication and viral infection of vB_Pae_PLY. No potential antibiotic resistance, virulence, or tRNA genes were identified. Information about functional annotation of the 64 CDS and their respective proteins is detailed in Table 1.

The genomic structure of vB_Pae_PLY. The first circles represent the 64 coding DNA sequences (CDS) of the phage genome. The second circle shows GC content. The third circle shows the GC skew
Phage taxonomy and phylogeny analysis
To classify vB_Pae_PLY, a viral proteomics tree was created. The analysis showed that vB_Pae_PLY belonged to the family Autographiviridae, which was consistent with the results observed via TEM (Fig. 5a). A phylogenetic tree based on 349 (including vB_Pae_PLY) genome sequences of all Pseudomonadota phages of the family Autophagoviridae depicted that vB_Pae_PLY was most closely related to the five phages (phage PAXYB1, phage MPK6, phage RLP, phage LUZ19, and phage DL62) of Phikmvvirus(Fig. 5b). Therefore, the phage vB_Pae_PLY was assigned to the genus Phikmvvirus of the subfamily Krylovinae, the family Autographiviridae, without order classification.

Phage taxonomy and phylogeny analysis. (a) Viral proteomic tree; (b) Phylogenetic tree based on the 349 genome sequences of Pseudomonadota phages of the family Autophagoviridae
The adsorption receptor of vB_Pae_PLY
Phage adsorption to the bacterial surface is the first and most crucial step in the phage infection process. LPS and outer membrane proteins located on the surface of Gram-negative bacteria can both serve as phage receptors. Proteinase K and NaIO4 were used to disrupt the bacterial surface component to identify the attachment sites of vB_Pae_PLY. PAO1 showed a significant decrease in phage adsorption following NaIO4 treatment (P < 0.01), but no observable changes were found after the proteinase K treatment (Fig. 6a). As periodate destroys the structure of LPS and protease K destroys outer membrane proteins, it could be inferred that LPS is an adsorption receptor for vB_Pae_PLY. This result was further confirmed by the LPS adsorption assay, in which phage adsorption to P. aeruginosa increased when LPS was added to the reaction system (Fig. 6b).

Identification of LPS as an important absorption receptor of vB_Pae_PLY. (a) NaIO4 treatment significantly reduced the adsorption of vB_Pae_PLY; (b) LPS adsorption assay. The adsorption rate was increased in the LPS-added group compared to that in the control group. **P < 0.01, ns, no significance
The adsorption rate and bactericidal activity of vB_Pae_PLY after lasR knockout
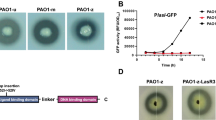
Transcriptome analysis showed that lasR expression decreased significantly, and QS was down-regulated, indicating that lasR was successfully knocked out (Figure S1). The study further investigated the influence of the Las QS system on phage infection. The results of the spot assay showed that lasR deletion reduced the clarity of clear patches formed by vB_Pae_PLY (Fig. 7a). The adsorption rates of phages were compared, which revealed that PaΔlasR showed a significant decrease in phage adsorption compared to wild-type PAO1 (P < 0.05) (Fig. 7b). The results suggested that lasR knockout may influence phage sensitivity by reducing phage adsorption to P. aeruginosa.

Phage sensitivity assay. (a) Ten-fold serial dilutions of the phage vB_Pae_PLY plated on PAO1 and lasR mutants PaΔlasR; (b) Comparison of adsorption rates of the phage vB_Pae_PLY to PAO1 and PAO1ΔlasR; (c) Time killing effect in the presence or absence of phage at an MOI of 10. *P < 0.05
Additionally, colony-counting results showed that compared to wild-type PAO1, the bactericidal effect of vB_Pae_PLY against PaΔlasR was reduced (Fig. 7c). At 4 h and 8 h, the colony count in the PaΔlasR group was higher than that in the wild group, and the phage resistant bacteria load of PaΔlasR was higher than that in the wild group. At 12 h, there was little difference in bacterial load between the two groups. These results showed that lasR QS inhibition weakened the bactericidal efficiency of the phage vB_Pae_PLY.
The expression of LPS and T4P synthesis genes after lasR knockout
A differential gene expression experimen was performed by RT-qPCR to gain insights into the mechanism of changes in the sensitivity of PaΔlasR to the phage. The expression level of LPS synthesis gene galU was significantly decreased after lasR knockout. In addition, T4P synthesis related genes (including pilA, pilB, pilC and pilD, pilQ and pilV, pilW and pilY) were also markedly down-regulated (Fig. 8). As LPS is a key adsorption receptor for phages and T4P is important in phage adsorption, the down-regulation of LPS and T4P related genes caused by lasR knockout may be the main reason for the decreased phage sensitivity.

lasR knockout down-regulated the expression of phage absorption-related genes. The reference gene was rpsL. *P < 0.05, **P < 0.01, and ***P < 0.001
Discussion
Studies have shown that in some Gram-negative bacteria like Vibrio and Escherichia coli (E. coli), QS regulated phage resistance by down-regulating phage receptors on the cell surface. M Mozammel et al. indicated that QS may mediate phage resistance by reducing or eliminating O antigen expression in Vibrio cholerae [30]. Down-regulation of the outer membrane protein K (OmpK) receptor mediated by QS in Vibrio anguillarum reduced phage adsorption [17]. In E. coli, the QS system reduced phage receptors [16, 31]. Additionally, the study by Xuan et al. revealed that N-acyl homoserine lactones (AHLs) protected Shewanella baltica from phage infection by decreasing LPS-mediated phage adsorption [32]. The above studies all hypothesized that QS negatively regulates the expression of receptors required for phage infection. However, while the phenomena and mechanisms of QS-controlled phage resistance have been observed in these bacteria, it has not been clear whether QS has similar effects in other bacteria. Interestingly, the present study found that the knockout of lasR in P. aeruginosa PAO1 decreased the phage sensitivity and adsorption (Fig. 7), indicating that lasR positively regulated phage infection. This is consistent with previous observations in P. aeruginosa [18, 33]. This study demonstrated that lasR promotes phage infection in P. aeruginosa.
Phage adsorption to the bacterial surface is the first and most critical step in phage infection [34, 35]. Molecules on the bacterial surface, such as LPS, pili, peptidoglycan components, and outer membrane proteins, can all be attachment sites for phage tails [35,36,37,38]. LPS is related to virulence in various Gram-negative bacteria, and is also the receptor of many phages [39,40,41,42]. Some LPS-specific phages against P. aeruginosa, and phage-resistant strains generated by mutations in LPS biosynthetic genes have been described in the literature [43,44,45,46,47]. T4P plays a central role in the expression of many phenotypes including motility, multicellular behavior, sensitivity to phages, natural genetic transformation, and adhesion [48, 49]. Studies show that many P. aeruginosa phages utilize pili as the primary receptor to infect cells, and mutations or inhibition of T4P synthesis may reduce the adsorption [18, 40, 41, 46, 47]. Downregulation of pilA, pilB and pilQ genes reduced T4P-mediated phage adsorption to protect P. aeruginosa from phage infection [41, 50].
RT-qPCR analysis in the present study revealed that galU and T4P-related genes (pilA, pilB, pilC and pilD, pilQ and pilV, pilW and pilY) were significantly decreased in the PaΔlasR (Fig. 8). LPS is composed of O antigen, core oligosaccharide, and lipid A. galU encodes UDP-glucose pyrophosphorylase (GalU), and was found to be involved in the synthesis of the core region of P. aeruginosa LPS [43, 51]. The current study demonstrates that the phage vB_Pae_PLY utilizes surface LPS as the receptor for adsorption (Fig. 6). Thus, the reduced phage sensitivity in PaΔlasR can be largely attributed to the impaired LPS synthesis. In addition, pili also play an important role in phage infection; therefore, downregulation of T4P may also be another reason for reduced phage sensitivity. LPS and T4P are important virulence factors for the opportunistic pathogen P. aeruginosa. On the other hand, serving as phage receptors, LPS and T4P facilitate phage infection with their synthesis being positively regulated by QS. Therefore, lasR QS inhibition will affect the synthesis of bacterial LPS and T4P, resulting in reduced phage adsorption with LPS and T4P as receptors, and weaken the bactericidal effect of phages to a certain extent.
Conclusions
In conclusion, this study demonstrates that lasR may promote phage infection by positively regulating the biosynthesis of LPS and T4P. Disrupting lasR expression leads to decreased phage sensitivity. Given the complex and multifaceted role of QS in host-phage interactions, the future research is needed to uncover the various mechanisms by which QS participates in phage infection.
Data availability
The complete genome sequence of phage vB_Pae_PLY presented in this study is openly available in [GenBank, OR689712].
Abbreviations
- Pseudomonas aeruginosa :
-
P. aeruginosa
- QS:
-
Quorum sensing
- RT-qPCR:
-
Real-Time quantitative polymerase chain reaction
- LPS:
-
Lipopolysaccharide
- T4P:
-
Type IV pili-related
- AHL:
-
N-acylhomoserine lactones
- 3-oxo-C12-HSL:
-
N -(3-oxo-dodecanoyl)- L -homoserine lactone
- C4-HSL:
-
N -(butanoyl)- L -homoserine lactone
- HHQ:
-
2-heptyl-4-hydroxyquinoline
- PQS:
-
2-heptyl-3-hydroxy-4(1 H)-quinolone
- IQS:
-
2-(2-hydroxyphenyl)-thiazole-4-carbaldehyde
- LB:
-
Luria-Bertani
- Car:
-
Carbenicillin
- Gen:
-
Gentamicin
- PFU:
-
Plaque-forming unit
- CFU:
-
Colony-forming unit
- VFDB:
-
Virulence Factor Database
- CARD:
-
Comprehensive Antibiotic Resistance Database
- PCR:
-
Polymerase chain reaction
- PBS:
-
Phosphate-buffered saline
- MOI:
-
Multiplicity of infection
- TEM:
-
Transmission electron microscopy
- CDS:
-
Coding DNA sequences
- ICTV:
-
International Committee on Taxonomy of Viruses
- OmpK:
-
Outer membrane protein K
- GalU:
-
UDP-glucose pyrophosphorylase
References
Wang J, Wang C, Yu HB, Dela Ahator S, Wu X, Lv S, et al. Bacterial quorum-sensing signal IQS induces host cell apoptosis by targeting POT1-p53 signalling pathway. Cell Microbiol. 2019;21(10):e13076. https://doi.org/10.1111/cmi.13076.
Montagut EJ, Marco MP. Biological and clinical significance of quorum sensing alkylquinolones: current analytical and bioanalytical methods for their quantification. Anal Bioanal Chem. 2021;413(18):4599–618. https://doi.org/10.1007/s00216-021-03356-x.
Rather MA, Saha D, Bhuyan S, Jha AN, Mandal M. Quorum quenching: a Drug Discovery Approach against Pseudomonas aeruginosa. Microbiol Res. 2022;264:127173. https://doi.org/10.1016/j.micres.2022.127173.
Giallonardi G, Letizia M, Mellini M, Frangipani E, Halliday N, Heeb S, et al. Alkyl-Quinolone-dependent quorum sensing controls prophage-mediated autolysis in Pseudomonas aeruginosa colony biofilms. Front Cell Infect Microbiol. 2023;13:1183681. https://doi.org/10.3389/fcimb.2023.1183681.
Lee J, Zhang L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell. 2015;6(1):26–41. https://doi.org/10.1007/s13238-014-0100-x.
Chadha J, Harjai K, Chhibber S. Revisiting the virulence hallmarks of Pseudomonas aeruginosa: a chronicle through the perspective of quorum sensing. Environ Microbiol. 2022;24(6):2630–56. https://doi.org/10.1111/1462-2920.15784.
Schroven K, Putzeys L, Swinnen AL, Hendrix H, Paeshuyse J, Lavigne R. The phage-encoded protein PIT2 impacts Pseudomonas aeruginosa quorum sensing by direct interaction with LasR. iScience. 2023;26(10):107745. https://doi.org/10.1016/j.isci.2023.107745.
Wang Y, Dai J, Wang X, Wang Y, Tang F. Mechanisms of interactions between bacteria and bacteriophage mediate by quorum sensing systems. Appl Microbiol Biotechnol. 2022;106(7):2299–310. https://doi.org/10.1007/s00253-022-11866-6.
Rutherford ST, Bassler BL. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med. 2012;2(11). https://doi.org/10.1101/cshperspect.a012427.
Li P, Lin H, Mi Z, Xing S, Tong Y, Wang J. Screening of Polyvalent phage-resistant Escherichia coli strains based on phage receptor analysis. Front Microbiol. 2019;10:850. https://doi.org/10.3389/fmicb.2019.00850.
Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat Rev Microbiol. 2010;8(5):317–27. https://doi.org/10.1038/nrmicro2315.
Ahator SD, Sagar S, Zhu M, Wang J, Zhang LH. Nutrient availability and phage exposure alter the Quorum-Sensing and CRISPR-Cas-Controlled Population dynamics of Pseudomonas aeruginosa. mSystems. 2022;7(4):e0009222. https://doi.org/10.1128/msystems.00092-22.
De Smet J, Hendrix H, Blasdel BG, Danis-Wlodarczyk K, Lavigne R. Pseudomonas predators: understanding and exploiting phage-host interactions. Nat Rev Microbiol. 2017;15(9):517–30. https://doi.org/10.1038/nrmicro.2017.61.
Høyland-Kroghsbo NM, Paczkowski J, Mukherjee S, Broniewski J, Westra E, Bondy-Denomy J, et al. Quorum sensing controls the Pseudomonas aeruginosa CRISPR-Cas adaptive immune system. Proc Natl Acad Sci U S A. 2017;114(1):131–5. https://doi.org/10.1073/pnas.1617415113.
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–12. https://doi.org/10.1126/science.1138140.
Høyland-Kroghsbo NM, Maerkedahl RB, Svenningsen SL. A quorum-sensing-induced bacteriophage defense mechanism. mBio. 2013;4(1):e00362–12. https://doi.org/10.1128/mBio.00362-12.
Tan D, Svenningsen SL, Middelboe M. Quorum sensing determines the choice of Antiphage Defense Strategy in Vibrio anguillarum. mBio. 2015;6(3):e00627. https://doi.org/10.1128/mBio.00627-15.
Xuan G, Lin H, Tan L, Zhao G, Wang J. Quorum sensing promotes phage infection in Pseudomonas aeruginosa PAO1. mBio. 2022;13(1):e0317421. https://doi.org/10.1128/mbio.03174-21.
Liu Y, Zhao Y, Qian C, Huang Z, Feng L, Chen L, et al. Study of combined effect of bacteriophage vB3530 and chlorhexidine on the inactivation of Pseudomonas aeruginosa. BMC Microbiol. 2023;23(1):256. https://doi.org/10.1186/s12866-023-02976-w.
Fang Q, Feng Y, McNally A, Zong Z. Characterization of phage resistance and phages capable of intestinal decolonization of carbapenem-resistant Klebsiella pneumoniae in mice. Commun Biol. 2022;5(1):48. https://doi.org/10.1038/s42003-022-03001-y.
Xuan G, Kong J, Wang Y, Lin H, Wang J. Characterization of the newly isolated Pseudomonas phage vB_Pae_LC3I3. Virus Res. 2023;323:198978. https://doi.org/10.1016/j.virusres.2022.198978.
Grant JR, Enns E, Marinier E, Mandal A, Herman EK, Chen CY, et al. Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023;51(W1):W484–92. https://doi.org/10.1093/nar/gkad326.
Li E, Yin Z, Ma Y, Li H, Lin W, Wei X, et al. Identification and molecular characterization of bacteriophage phiAxp-2 of Achromobacter xylosoxidans. Sci Rep. 2016;6:34300. https://doi.org/10.1038/srep34300.
Kiljunen S, Datta N, Dentovskaya SV, Anisimov AP, Knirel YA, Bengoechea JA, et al. Identification of the lipopolysaccharide core of Yersinia pestis and Yersinia pseudotuberculosis as the receptor for bacteriophage φA1122. J Bacteriol. 2011;193(18):4963–72. https://doi.org/10.1128/jb.00339-11.
Zhang Y, Wang L, Chen L, Zhu P, Huang N, Chen T, et al. Novel insight of transcription factor PtrA on pathogenicity and Carbapenems Resistance in Pseudomonas aeruginosa. Infect Drug Resist. 2022;15:4213–27. https://doi.org/10.2147/idr.S371597.
Jeon J, D’Souza R, Pinto N, Ryu CM, Park J, Yong D, et al. Characterization and complete genome sequence analysis of two myoviral bacteriophages infecting clinical carbapenem-resistant Acinetobacter baumannii isolates. J Appl Microbiol. 2016;121(1):68–77. https://doi.org/10.1111/jam.13134.
Li J, Yan B, He B, Li L, Zhou X, Wu N, et al. Development of phage resistance in multidrug-resistant Klebsiella pneumoniae is associated with reduced virulence: a case report of a personalised phage therapy. Clin Microbiol Infect. 2023. https://doi.org/10.1016/j.cmi.2023.08.022.
Liu Y, Wang Y, Kong J, Jiang X, Han Y, Feng L, et al. An effective antimicrobial strategy of colistin combined with the Chinese herbal medicine shikonin against colistin-resistant Escherichia coli. Microbiol Spectr. 2023;e0145923. https://doi.org/10.1128/spectrum.01459-23.
Adriaenssens EM, Sullivan MB, Knezevic P, van Zyl LJ, Sarkar BL, Dutilh BE, et al. Taxonomy of prokaryotic viruses: 2018–2019 update from the ICTV bacterial and archaeal viruses Subcommittee. Arch Virol. 2020;165(5):1253–60. https://doi.org/10.1007/s00705-020-04577-8.
Hoque MM, Naser IB, Bari SM, Zhu J, Mekalanos JJ, Faruque SM. Quorum Regulated Resistance of Vibrio cholerae against Environmental bacteriophages. Sci Rep. 2016;6:37956. https://doi.org/10.1038/srep37956.
Ding Y, Zhang D, Zhao X, Tan W, Zheng X, Zhang Q, et al. Autoinducer-2-mediated quorum-sensing system resists T4 phage infection in Escherichia coli. J Basic Microbiol. 2021;61(12):1113–23. https://doi.org/10.1002/jobm.202100344.
Xuan G, Tan L, Yang Y, Kong J, Lin H, Wang J. Quorum sensing autoinducers AHLs protect Shewanella baltica against phage infection. Int J Food Microbiol. 2023;403:110304. https://doi.org/10.1016/j.ijfoodmicro.2023.110304.
Broniewski JM, Chisnall MAW, Høyland-Kroghsbo NM, Buckling A, Westra ER. The effect of Quorum sensing inhibitors on the evolution of CRISPR-based phage immunity in Pseudomonas aeruginosa. Isme j. 2021;15(8):2465–73. https://doi.org/10.1038/s41396-021-00946-6.
Nobrega FL, Vlot M, de Jonge PA, Dreesens LL, Beaumont HJE, Lavigne R, et al. Targeting mechanisms of tailed bacteriophages. Nat Rev Microbiol. 2018;16(12):760–73. https://doi.org/10.1038/s41579-018-0070-8.
Dunne M, Hupfeld M, Klumpp J, Loessner MJ. Molecular basis of bacterial host interactions by Gram-positive targeting bacteriophages. Viruses. 2018;10(8). https://doi.org/10.3390/v10080397.
Donlan RM. Preventing biofilms of clinically relevant organisms using bacteriophage. Trends Microbiol. 2009;17(2):66–72. https://doi.org/10.1016/j.tim.2008.11.002.
Dowah ASA, Clokie MRJ. Review of the nature, diversity and structure of bacteriophage receptor binding proteins that target Gram-positive bacteria. Biophys Rev. 2018;10(2):535–42. https://doi.org/10.1007/s12551-017-0382-3.
Bertozzi Silva J, Storms Z, Sauvageau D. Host receptors for bacteriophage adsorption. FEMS Microbiol Lett. 2016;363(4). https://doi.org/10.1093/femsle/fnw002.
Harvey H, Bondy-Denomy J, Marquis H, Sztanko KM, Davidson AR, Burrows LL. Pseudomonas aeruginosa defends against phages through type IV pilus glycosylation. Nat Microbiol. 2018;3(1):47–52. https://doi.org/10.1038/s41564-017-0061-y.
McCutcheon JG, Peters DL, Dennis JJ. Identification and characterization of type IV Pili as the Cellular receptor of broad host Range Stenotrophomonas maltophilia bacteriophages DLP1 and DLP2. Viruses. 2018;10(6). https://doi.org/10.3390/v10060338.
Xuan G, Lin H, Wang J. Expression of a phage-encoded Gp21 protein protects Pseudomonas aeruginosa against phage infection. J Virol. 2022;96(5):e0176921. https://doi.org/10.1128/jvi.01769-21.
Testa S, Berger S, Piccardi P, Oechslin F, Resch G, Mitri S. Spatial structure affects phage efficacy in infecting dual-strain biofilms of Pseudomonas aeruginosa. Commun Biol. 2019;2:405. https://doi.org/10.1038/s42003-019-0633-x.
Dean CR, Goldberg JB. Pseudomonas aeruginosa galU is required for a complete lipopolysaccharide core and repairs a secondary mutation in a PA103 (serogroup O11) wbpM mutant. FEMS Microbiol Lett. 2002;210(2):277–83. https://doi.org/10.1111/j.1574-6968.2002.tb11193.x.
Li L, Pan X, Cui X, Sun Q, Yang X, Yang H. Characterization of Pseudomonas aeruginosa phage K5 genome and identification of its receptor related genes. J Basic Microbiol. 2016;56(12):1344–53. https://doi.org/10.1002/jobm.201600116.
Oechslin F, Piccardi P, Mancini S, Gabard J, Moreillon P, Entenza JM, et al. Synergistic Interaction between Phage Therapy and Antibiotics clears Pseudomonas Aeruginosa infection in Endocarditis and reduces virulence. J Infect Dis. 2017;215(5):703–12. https://doi.org/10.1093/infdis/jiw632.
Pires DP, Dötsch A, Anderson EM, Hao Y, Khursigara CM, Lam JS, et al. A Genotypic Analysis of Five P. aeruginosa strains after Biofilm infection by phages targeting different cell surface receptors. Front Microbiol. 2017;8:1229. https://doi.org/10.3389/fmicb.2017.01229.
Li N, Zeng Y, Wang M, Bao R, Chen Y, Li X, et al. Characterization of Phage Resistance and their impacts on bacterial fitness in Pseudomonas aeruginosa. Microbiol Spectr. 2022;10(5):e0207222. https://doi.org/10.1128/spectrum.02072-22.
Piepenbrink KH, Sundberg EJ. Motility and adhesion through type IV pili in Gram-positive bacteria. Biochem Soc Trans. 2016;44(6):1659–66. https://doi.org/10.1042/bst20160221.
Craig L, Forest KT, Maier B, Type. IV pili: dynamics, biophysics and functional consequences. Nat Rev Microbiol. 2019;17(7):429–40. https://doi.org/10.1038/s41579-019-0195-4.
Xuan G, Dou Q, Kong J, Lin H, Wang J. Pseudomonas aeruginosa resists phage infection via eavesdropping on Indole Signaling. Microbiol Spectr. 2023;11(1):e0391122. https://doi.org/10.1128/spectrum.03911-22.
Jiang SS, Lin TY, Wang WB, Liu MC, Hsueh PR, Liaw SJ. Characterization of UDP-glucose dehydrogenase and UDP-glucose pyrophosphorylase mutants of Proteus mirabilis: defectiveness in polymyxin B resistance, swarming, and virulence. Antimicrob Agents Chemother. 2010;54(5):2000–9. https://doi.org/10.1128/aac.01384-09.
Funding
This work was supported by the Health Department of Zhejiang Province of the People’s Republic of China, No. 2022KY895; the research grants from the National Natural Science Foundation of China, No. 82172328; and the Key Laboratory of Clinical Laboratory Diagnosis and Translational Research of Zhejiang Province, No. 2022E10022.
Author information
Authors and Affiliations
Contributions
YL and ZCY performed the experiments, generated figures, analyzed the data, and drafted the manuscript under the supervision of QW and TLZ. ZZM, MRT, HCC, CRQ, and WLZ conducted the data analysis. The research plan for this project was conceived based on several rounds of discussions among all co-authors. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All the experimental methods were performed in accordance with approved guidelines and regulations. The First Affiliated Hospital of Wenzhou Medical University’s ethics committee authorized each investigative protocol used in this study. Informed consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, Y., Yao, Z., Mao, Z. et al. Quorum sensing gene lasR promotes phage vB_Pae_PLY infection in Pseudomonas aeruginosa. BMC Microbiol 24, 207 (2024). https://doi.org/10.1186/s12866-024-03349-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-024-03349-7