
Abstract
Background
The urinary tract harbors unique microbial communities that play important roles in urogenital health and disease. Dogs naturally suffer from several of the same urological disorders as humans (e.g., urinary tract infections, neoplasia, urolithiasis) and represent a valuable translational model for studying the role of urinary microbiota in various disease states. Urine collection technique represents a critical component of urinary microbiota research study design. However, the impact of collection method on the characterization of the canine urinary microbiota remains unknown. Therefore, the objective of this study was to determine whether urine collection technique alters the microbial populations detected in canine urine samples. Urine was collected from asymptomatic dogs by both cystocentesis and midstream voiding. Microbial DNA was isolated from each sample and submitted for amplicon sequencing of the V4 region of the bacterial 16 S rRNA gene, followed by analyses to compare microbial diversity and composition between urine collection techniques.
Results
Samples collected via midstream voiding exhibited significantly higher sequence read counts (P = .036) and observed richness (P = .0024) than cystocentesis urine. Bray Curtis and Unweighted UniFrac measures of beta diversity showed distinct differences in microbial composition by collection method (P = .0050, R2 = 0.06 and P = .010, R2 = 0.07, respectively). Seven taxa were identified as differentially abundant between groups. Pasteurellaceae, Haemophilus, Friedmanniella, two variants of Streptococcus, and Fusobacterium were over-represented in voided urine, while a greater abundance of Burkholderia-Caballeronia-Paraburkholderia characterized cystocentesis samples. Analyses were performed at five thresholds for minimum sequence depth and using three data normalization strategies to validate results; patterns of alpha and beta diversity remained consistent regardless of minimum read count requirements or normalization method.
Conclusion
Microbial composition differs in canine urine samples collected via cystocentesis as compared to those collected via midstream voiding. Future researchers should select a single urine collection method based on the biological question of interest when designing canine urinary microbiota studies. Additionally, the authors suggest caution when interpreting results across studies that did not utilize identical urine collection methods.
Similar content being viewed by others


Background
Emerging research has established that the urinary tract, previously thought to be sterile, hosts a unique and diverse microbial ecosystem [1,2,3,4,5,6]. Urinary microbial communities play essential roles in urogenital health and disease, and disorders such as urinary incontinence, urolithiasis, and bladder neoplasia have been linked to alterations in human urinary microbiota (UM) [6,7,8,9,10,11,12]. Further research investigating the UM is critical to improving our understanding of host-microbe relationships within the urinary tract and exposing novel treatment and preventative strategies for disorders associated with urinary dysbiosis.
Microbiota analyses of the urinary tract present several unique challenges for researchers. Most notably, urine possesses low biomass, which creates vulnerability to contamination and technical biases. To address these challenges, UM studies require careful specimen collection, handling, and bioinformatic processing. Sample collection technique represents a particularly critical component of UM study design, as biological sources of contamination vary by urine collection method. Urine samples are typically collected by either midstream voiding (i.e., free catch), urethral catheterization, suprapubic/antepubic aspiration (i.e., cystocentesis), or cystoscope evacuation. In humans, sample collection technique alters UM composition [1, 10, 13,14,15,16,17]. Voided samples risk contamination with urethral, genital, and dermal microbiota, whereas cystocentesis and urethral catheterization are thought to better reflect the microbiota of bladder urine. In fact, a recent consensus statement recommends differentiating urinary tract microbiota terminology, reserving the designation “urinary bladder” for samples collected directly from the bladder (urethral catheterization, cystocentesis, or cystoscopic collection), and using the term “urogenital” for voided samples [12].
The dog is a valuable model for urogenital and microbiota-related research, and investigation of the canine UM might inform disease pathology relevant to both canine and human health, such as urolithiasis, urinary tract infections, neoplasia, and neurogenic bladder [18,19,20,21]. The presence of urinary microbial communities in dogs is also well-established [22,23,24,25,26], yet the impact of urine collection method on microbiota composition in dogs remains unknown. Past studies examining the canine UM have included urine collected by either cystocentesis or midstream voiding, which are two of the most common methods of canine urine collection performed in clinical settings [22,23,24,25,26]. The microbiota of canine urine collected via cystocentesis shares more similarities with the microbiota of genital swabs than rectal swabs from the same animal [22]. However, the effect of urine collection technique on canine UM detection remains unknown. This is a critical issue given that urethral, genital, or dermal microbial contamination of voided urine might alter UM composition in dogs.
Improved understanding of how urine sample collection affects these microbial communities is essential for the design and interpretation of canine UM studies. Therefore, the primary objective of this study was to characterize the impact of urine collection method on microbial abundance, diversity, and composition in canine urine samples. A secondary objective was to determine how specific aspects of the analysis pipeline, including minimum read count (i.e., sequencing depth) and data normalization method, affect results.
Methods
Patient selection
Spayed or neutered dogs between the ages of 1 and 7 years old were actively recruited through an email announcement about the study to employees at the University of Minnesota’s College of Veterinary Medicine until a total of 10 male and 10 female dogs meeting the study inclusion criteria were enrolled. Dogs with a diagnosis of lower urinary tract disease or clinical signs localizing to the lower urinary tract were excluded. Clinical signs of lower urinary tract disease were defined as gross hematuria, stranguria, pollakiuria, or dysuria. Dogs with active dermatologic disease or visible dermatologic lesions in the perivulvar, preputial, or inguinal areas were not included. Dogs with a diagnosis or clinical signs of chronic or acute (within 3 weeks of study enrollment) gastrointestinal disease were also excluded. Clinical signs of gastrointestinal distress included vomiting, diarrhea, hematochezia, borborygmus, hyporexia, anorexia, or abdominal bloating. Finally, dogs that received antimicrobial or immunosuppressive therapies within 3 months of study enrollment were excluded. Signalment, body weight, body condition score (BCS, 1–9 scale), hair length, presence of dermatologic lesions, medical history, diet, medication history, urinalysis results, and urine culture results were recorded for each study participant. Hair length was categorized as short (< 1 inch), medium (1–2 inches), or long (> 2 inches).
Sample collection
For each participant, antepubic cystocentesis was performed first, followed by collection of voided urine. Skin was cleansed with 70% isopropyl alcohol prior to cystocentesis. Five mL of urine was collected via cystocentesis, and the collection needle was exchanged for a sterile needle prior to transfer to a sterile tube. A minimum of 7 mL of midstream, voided urine was collected in a sterile cup by a veterinarian or certified veterinary technician within 6 h of cystocentesis. Samples were refrigerated immediately after collection and transported to a -80˚ C freezer within 2 h of collection. The time between cystocentesis and voided urine collection was documented for each dog. If a dog voided immediately after cystocentesis, the interval was recorded as 0.1 h.
An aliquot of the voided urine sample ranging from 1 to 6 mL was submitted for urinalysis. Dogs with pyuria (> 5 white blood cells/hpf) or cytologic bacteriuria were excluded. Urine specific gravity (USG) was measured using a digital veterinary refractometer (MISCO Palm Abbe, Solon, OH), and urine pH was determined using a urine dipstick chemistry test (Siemens MultiStix, Malvern, PA). For dogs with a USG reported as > 1.045, the USG was recorded as 1.045 for analyses. Aerobic bacterial culture was also performed from voided urine from each dog. Approximately 0.5 mL of urine was gently mixed, and a 1 µL inoculation loop (Globe Scientific, Mahwah, New Jersey) was used to perform a streak plate inoculation of a Blood Agar Plate (Hardy Diagnostics, Santa Maria, California), followed by incubation at 37˚ C for 48 h. Significant bacterial growth was defined as greater than or equal to 105 CFU/mL according to previous guidelines [27]. Thus, samples with bacterial growth ≥ 105 CFU/mL after 48 h of incubation were excluded.
DNA isolation and amplicon sequencing
Five mL of urine from each collection method were reserved for bacterial 16 S rRNA gene amplification and sequencing. Prior to DNA extraction, urine was pelleted by centrifugation at 3,000 rpm for 15 min [28]. The DNeasy PowerSoil Pro Kit (QIAGEN, Hilden, Germany) was used to extract microbial DNA from the urine in accordance with manufacturer instructions. Two negative controls were included, which consisted only of DNA extraction reagents. The ZymoBIOMICS Microbial Community Standard (Zymo Research, Irvine, California) was used as a positive control. This is a mock bacterial community comprised of eight organisms: Pseudomonas aeruginosa, Escherichia coli, Salmonella enterica, Lactobacillus fermentum, Enterococcus faecalis, Staphylococcus aureus, Listeria monocytogenes, and Bacillus subtilis.
DNA concentration and purity was determined using Quanti-iT PicoGreen dsDNA Assay kit (Invitrogen, Waltham, MA) and NanoDrop spectrophotometry (Thermo Fisher Scientific, Waltham, MA). The V4 region of the bacterial 16 S rRNA gene was amplified using primers 515 F (GTGCCAGCMGCCGCGGTAA) and 806R (GGACTACHVGGGTWTCTAAT) [29]. Amplicon sequencing of all clinical samples, negative controls, and the positive control were performed on the MiSeq sequencing platform, targeting 2 × 300 bp paired-end reads, v3 chemistry (Illumina, San Diego, CA). DNA extractions and library preparation were performed in a single batch. DNA extraction, library preparation, PCR amplification, and 16 S rRNA short amplicon sequencing were performed at the University of Minnesota Genomics Center.
Bioinformatic processing
Raw, paired-end sequence reads were processed using Cutadapt [30] for primer removal, followed by QIIME2 (v. 2020.8) [31] and DADA2 [32] for the removal of low-quality base pairs, sequence truncation, and forward and reverse read merger. Taxonomic assignment of high-resolution amplicon sequence variant (ASV) outputs was performed using the Silva reference database (v. 138) [33]. Chimeras, mitochondria, chloroplasts, ASVs with unassigned taxonomy, and ASVs present in only a single sample were removed from the dataset prior to downstream analysis. Sequences represented in less than 10 total reads across all samples were also removed, as previously described [8, 34]. The decontam package (v. 1.10.0) within the R statistical software was used to identify contaminant taxa using the prevalence method with a threshold of 0.5 [35]. Features identified as putative contaminants via decontam were removed from the dataset prior to downstream analysis. A threshold of 300 sequence reads was set for the primary analyses, and any sample with less than 300 reads was removed from analysis [24]. Only data from dogs with paired cystocentesis and voided urine samples meeting these criteria were included in analyses. Sequence read counts were normalized via relative abundance transformation for the primary analyses.
Data analysis
Normality of continuous variables was determined using the Shapiro-Wilk test within the R statistical software (v. 4.0.2). The vegan (v. 2.5-7) [36] and phyloseq (v. 1.34.0) [37] packages were used to perform alpha and beta diversity analyses. Alpha diversity measures included Shannon diversity index, inverse Simpson diversity index, observed richness, and Pielou’s evenness. Alpha diversity was compared between experimental groups using Wilcoxon signed-rank test (analysis by collection method) or Wilcoxon rank-sum tests (analysis by sex). Measures of beta diversity included Bray Curtis dissimilarity, Weighted UniFrac, and Unweighted UniFrac. Statistical differences in microbial composition between groups (urine collection method and sex) was assessed using permutational multivariate analysis of variance (PERMANOVA) with 1000 permutations. A two-way PERMANOVA (adonis2 function in R vegan package) was also performed to assess for interactions between urine collection method and sex.
Identification of differentially abundant organisms between experimental groups was performed using indicator species analysis [38] in the R labdsv package (v.2.0–1) [39]. Indicator species analysis assigns an indicator value (IV) to each ASV by calculating the product of the relative frequency and relative abundance of an individual taxon in an experimental group [38]. IVs range from 0 to 1, with higher values indicating that a taxon has higher abundance within samples and is more frequent across samples of a given experimental group. To be considered a significant differential feature by indicator species analysis, an IV of > 0.3 and a P value of < 0.05 was required, as previously described [40]. Indicator species analysis was performed between collection method, sex, and between sexes within a given collection method group. Organisms identified as differentially abundant by indicator species analysis were also evaluated with Wilcoxon signed-rank test (analysis by collection method) or Wilcoxon rank-sum test (analysis by sex) to compare overall abundance of organisms between experimental groups. Random forest analysis (classification mode; randomForest package v. 4.6–14) was also performed, and features with the highest discriminatory power between collection method groups using the mean decrease gini index were identified.
These data were further investigated using four additional sequence read thresholds (100, 700, 1000, 2000) and three normalization methods (performed at each sequence read threshold, including the primary threshold of 300). Normalization methods included rarefaction with random subsampling to the designated sequence read threshold (rrarefy function in R vegan package), relative abundance transformation, and negative binomial distribution using DESeq2 (v. 1.30.1) [41, 42]. A rarefaction curve was created (rarecurve function in R vegan package) to assess whether maximum diversity was achieved at the various sequence read thresholds.
Results
Study participants
Twenty-three dogs were recruited and screened for inclusion. Three dogs were excluded from the study during the sample collection phase. Two were excluded for behavioral causes, as the dogs’ temperaments prohibited urine collection, and the third was excluded due to visible inguinal pyoderma at the time of sample collection. Of the 20 dogs with urine samples collected, one female dog was later excluded due to contamination of her samples with the positive control during sample processing. The final study group comprised 19 dogs, including 10 neutered males and 9 spayed females. Indications for participant exclusions are summarized in Fig. 1.

Flow diagram showing the indications for exclusion of dogs and urine samples
Flow diagram illustrating the indications for excluding urine samples. A total of 13 dogs met study inclusion criteria and had paired cystocentesis and voided urine samples, each with at least 300 sequence reads.
Of these 19 dogs, mixed breeds were most common (n = 7), followed by Labrador Retrievers (n = 3). The following breeds were each represented once: Beagle, Boxer, Cavalier King Charles Spaniel, Doberman Pinscher, English Bulldog, German Shepherd Dog, Miniature Goldendoodle, Portuguese Water Dog, and Standard Poodle. Mean age of dogs was 3.6 years (± 1.7), and mean weight was 20.6 kg (± 10.5). BCS on a 1–9 scale ranged from 4.5 to 6.5 (median 5.5). Eight dogs had hair length classified as short and 11 as medium. No dogs had long hair. Mean urine pH was 7.3 (± 0.08), and USG ranged from 1.006 to 1.045 (median 1.042). No dogs had glucosuria or ketonuria observed on urinalysis, though 12 dogs exhibited microscopic hematuria (≤ 50 red blood cells/hpf for all but 1 dog). Two urine samples showed scant bacterial growth on aerobic culture (< 105 CFU/mL), and the remaining samples exhibited no observable bacterial growth. Twelve dogs (63%) voided immediately after cystocentesis, which was documented as 0.1 h. The time between cystocentesis and free catch collection ranged from 0.1 to 6 h (median 0.1 h).
Three dogs had an owner-reported history of suspected atopic dermatitis, but the disease was well-controlled with no pruritis or visible dermatologic lesions at the time of sample collection. A separate dog had mild erythema observed on the medial thighs and a history of well-controlled hypothyroidism, but no reported history of dermatologic disease or dermatologic lesions noted in the perivulvar or inguinal regions. All dogs consumed diets commercially formulated to meet the nutritional needs of adult dogs. Thirteen diets were represented across the 19 dogs, with four diets consumed by more than one dog and nine diets consumed by a single participant.
Filtering and initial data analysis
After initial filtering, eight organisms were identified as contaminants and removed from the dataset prior to downstream analysis (Additional file 1). Sequence reads across all samples ranged from 7 to 165,728 reads per sample (median 3649), comprising 414 unique ASVs. Samples collected via cystocentesis had a median of 1850 sequence reads, whereas samples collected via midstream voiding had a median of 61,947 sequence reads (P = .036). The voided sample exhibited a higher read count than the paired cystocentesis sample for 13 of 19 dogs. Sequence read counts from each urine sample, reported by dog and collection method, are displayed in Fig. 2.

Sequence read counts for paired cystocentesis and voided urine samples from 19 dogs
Sequence reads are reported for each urine sample, female dogs (F01 through F09) and male dogs (M01 through M10). Black bars represent cystocentesis samples, and gray bars represent midstream voided urine. Six dogs were excluded due to insufficient sequence reads (< 300) in at least one sample and are designated with an asterisk (*).
After filtration and putative contaminant removal, no reads were present in either negative control. In the positive control, each of the eight designated mock community members were identified, and no additional organisms were detected. Lowest assigned taxonomy of the mock community organisms was at the family level for Salmonella enterica, species level for Lactobacillus fermentum, and the remaining organisms were reported at the genus level. The abundance of organisms specified in the commercial product as compared to the abundance of organisms identified from 16 S rRNA gene amplicon sequencing is reported in Additional file 2.
Six of 38 urine samples (3 cystocentesis, 3 voided) had less than 300 sequence reads and were removed prior to downstream analysis (Figs. 1 and 2). All were from male dogs, and each sample with insufficient reads was from a different dog. Therefore, 6 dogs were removed for insufficient sequence reads in at least one sample, leaving 13 dogs (4 males, 9 females) that possessed paired samples meeting all study criteria.
Bacterial composition of urine samples
Ten bacterial phyla were represented across all urine samples. Firmicutes, Proteobacteria, and Actinobacteriota were the most common phyla observed across samples regardless of collection method, though relative abundance of specific phyla differed between groups (Fig. 3).

Bacterial phyla present in paired cystocentesis and voided urine samples from 13 dogs
Ten bacterial phyla were represented across samples. Urine collected via cystocentesis exhibited slightly higher proportions of Proteobacteria and Deinococcota and slightly lower proportions of Actinobacteriota as compared to voided urine.
Differences in alpha diversity were not detected between urine collected via cystocentesis and voided urine based on Shannon diversity index (P = .11, Fig. 4A) and inverse Simpson diversity index (P = .79, Fig. 4B). However, observed richness was higher in urine collected via midstream voiding than cystocentesis (P = .0024; Fig. 4C), whereas Pielou’s evenness was higher in urine collected via cystocentesis than midstream voiding (P = .011; Fig. 4D). Voided urine samples exhibited 333 unique ASVs (80% of total ASVs), meaning 333 ASVs were identified in at least one voided sample but in no samples collected via cystocentesis. However, only 10 ASVs (2% of total ASVs) were unique to cystocentesis urine samples and not present in voided urine.

Alpha diversity of paired cystocentesis and voided urine samples from 13 dogs
Cystocentesis samples are represented by closed circles, and voided samples are represented by open circles. Boxplot of alpha diversity as measured by the (A) Shannon diversity index (P = .11), (B) inverse Simpson diversity index (P = .79), (C) observed richness (P = .0024), and (D) Pielou’s evenness (P = .011). The boxes represent the 25th and 75th percentiles, and whiskers represent 1.5 times the interquartile range.
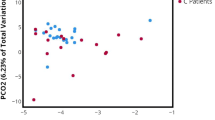
Beta diversity was assessed using three metrics: Bray Curtis dissimilarity matrix (Fig. 5A), Weighted UniFrac (Fig. 5B), and Unweighted UniFrac (Fig. 5C), and tested via PERMANOVA. Both Bray Curtis and Unweighted UniFrac metrics revealed minor but statistically significant differences in overall bacterial composition between cystocentesis and voided urine (P = .0050, R2 = 0.06 and P = .010, R2 = 0.07, respectively). Weighted UniFrac did not reveal a difference by collection method (P = .31, R2 = 0.04). No association was detected between the time interval between urine collections (Spearman’s correlation; rho = -0.17, P = .60) or hair length classification (P = .66) with Bray Curtis dissimilarity for an individual dog’s urine samples.

Beta diversity of paired cystocentesis and voided urine samples from 13 dogs
Principal coordinates analysis (PCoA) plot of beta diversity as measured by (A) Bray Curtis dissimilarity matrix (P = .0050, R2 = 0.06), (B) Weighted UniFrac (P = .31, R2 = 0.04), and (C) Unweighted UniFrac (P = .010, R2 = 0.07). The proportion of variation explained by each axis is listed in parentheses.
No measures of alpha diversity differed between male and female dogs (Shannon diversity index P = .72; inverse Simpson diversity index P = .89; observed richness P = .62; Pielou’s evenness P = .96). Beta diversity measures also did not differ by sex (Bray Curtis P = .75, R2 = 0.04; Weighted UniFrac P = .99, R2 = 0.02; Unweighted UniFrac P = .66, R2 = 0.04).
In the two-way PERMANOVA based on Bray Curtis dissimilarities, UM differed by collection method (P = .0060). However, there was no observed difference in UM by sex (P = .69), nor was there an interaction between sex and collection method (P = .53).
Differential abundance testing
Differential abundance of specific taxa between collection method group was assessed using indicator species analysis. Seven ASVs were identified (Table 1) with the criteria of IV > 0.3 and P < .05. Two of the differentially abundant ASVs exhibited IVs > 0.5: Burkholderia-Caballeronia-Paraburkholderia, which was over-represented in cystocentesis samples, and Pasteurellaceae, which characterized voided samples (Fig. 6). Note that this is a separate Burkholderia-Caballeronia-Paraburkholderia ASV than what was previously identified as a contaminant and removed from the dataset.

Top differentially abundant taxa between paired cystocentesis and voided urine samples from 13 dogs
Two amplicon sequence variants with differential abundance between cystocentesis and voided urine samples and an indicator value (IV) > 0.5 are visualized here. Cystocentesis samples are represented by closed circles, and midstream voided samples are represented by open circles. A pseudo-count of 0.01% relative abundance was added to each feature to allow log transformation visualization. Log transformation of relative abundance is displayed in boxplots for (A) Burkholderia-Caballeronia-Paraburkholderia (IV = 0.54, P for IV = 0.010, Wilcoxon signed-rank test P = .021), and (B) Pasteurellaceae (IV = 0.61, P for IV = 0.0070, Wilcoxon signed-rank test P = .014). The boxes represent the 25th and 75th percentiles, and whiskers represent 1.5 times the interquartile range.
Random forest analysis was performed to identify organisms with the highest discriminatory power when classifying samples by collection method (Fig. 7). The top 4 features identified via random forest analysis were also confirmed via indicator species analysis as differentially abundant between collection method groups (Table 1; Fig. 7).

Random forest analysis to detect discriminatory features when classifying canine urine as cystocentesis or voided
Random forest analysis using classification mode and mean decrease gini index was performed. The top 10 amplicon sequence variants with the highest discriminatory power between cystocentesis and voided urine samples are listed. The top four of these ten features were also identified as differentially abundant between collection technique groups using indicator species analysis. Taxa marked with an asterisk (*) indicate overrepresentation in the cystocentesis group designation. Taxa without an asterisk indicate overrepresentation in the voided group.
No organisms were identified as differentially abundant between males and females when evaluating all urine samples or when looking specifically at samples collected by cystocentesis. However, two organisms significantly differed between males and females when evaluating only voided urine samples. Schaalia canis was over-represented in voided urine from female dogs (IV = 0.56; P for IV = 0.039, Wilcoxon rank-sum P = .029), and Haemophilus was over-represented in voided urine from male dogs (IV = 0.69; P for IV = 0.031, Wilcoxon rank-sum P = .070).
Comparison of results using different sequence read thresholds and normalization methods
Urine samples and total dogs included with each described sequence read threshold (100, 300, 700, 1000, and 2000 sequence reads) are summarized in Additional file 3. Samples meeting the designated threshold ranged from 36 to 38 (95%) at the inclusion criterion of 100 reads to 22 of 38 (58%) at the inclusion criterion of 2000 reads. Additional file 4 shows a rarefaction curve indicating ASV richness at each sequence read threshold.
The findings from alpha and beta diversity testing between urine collection methods from analyses performed at each sequence read threshold and with each normalization method are summarized in Additional file 5. As compared to the primary analysis that used a minimum sequence read requirement of 300 and normalization via relative abundance transformation, patterns of alpha and beta diversity remained largely consistent across analyses despite different read requirements and normalization methods. For alpha diversity measures, differences in observed richness were detected between collection methods at all sequence read thresholds and normalization methods, and Shannon and inverse Simpson indices of alpha diversity were not found to differ by collection method for any sequence read thresholds, except 1000 reads. Pielou’s evenness differed by collection method using relative abundance and DESeq2 normalization for 3 of the 5 sequence read thresholds, including the primary analysis, but never for rarefaction analyses. For beta diversity measures, differences in Bray Curtis and Unweighted UniFrac were detected between collection methods in most (29 of 30) analyses. Maximum R2 values for Bray Curtis and Unweighted UniFrac across all analyses were 0.10 and 0.19, respectively, indicating that collection method explained less than 20% of UM variation. Weighted UniFrac did not identify statistically significant differences across any analysis. Finally, no differences in any measure of alpha or beta diversity were detected between male and female dogs using any sequence read cut-off or normalization method (Additional file 6).
Discussion
Our findings demonstrate that the canine UM differs in urine collected by cystocentesis as compared to urine collected by midstream voiding. Voided urine samples possessed significantly higher sequence reads and observed richness than urine collected via cystocentesis, and overall microbial composition differed between collection method based on two of three beta diversity measures. Several ASVs were identified as differentially abundant by collection method, most due to increased abundance in voided samples. No difference in measures of UM diversity were observed between male and female dogs in this study, though two organisms significantly differed by sex when evaluating only voided urine. When measures of UM diversity were compared using different minimum inclusion thresholds for sequence read counts and data normalization methods, overall significance patterns of alpha and beta diversity remained consistent. These findings confirm the presence of diverse microbial communities in canine urine and provide evidence that urine collection technique alters detection of UM composition in dogs.
In the current study, voided samples exhibited significantly higher sequence read counts as compared to urine collected by cystocentesis. While raw sequence read counts are not a direct reflection of sample biomass, we also observed that voided urine harbored roughly 30 times the number of unique organisms and exhibited increased observed richness when compared to cystocentesis samples. These findings likely reflect the presence of microbes from sources other than the bladder, such as the urethra, genital tract, or skin. In humans, there are distinctions in the UM between urine collection method, with voided urine samples often displaying increased abundance of urethral, genital, or peri-urethral/dermal microbes relative to urine collected via more direct bladder sampling (catheterization, cystocentesis, or cystoscopy) [1, 10, 13, 14, 16]. While we did not include microbiota analysis of urethral, genital, or dermal swabs to confirm the origin of specific microbes, all of the differentially abundant organisms over-represented in voided samples (Pasteurellaceae, Haemophilus, Friedmanniella, 2 variants of Streptococcus, and Fusobacterium), are components of the vaginal or skin microbiota of dogs, supporting the theory that dermal or urethro-genital contamination accounts for the increased abundance of these organisms in voided urine [22, 43,44,45,46,47,48]. Fusobacterium is also an abundant inhabitant of the canine gut microbiota [49, 50]. Taxa from the Pasteurellaceae family have been isolated from pharyngeal swabs in dogs [51], and Pasteurellaceae, Friedmanniella, and Streptococcus are also known components of the canine oral microbiota [52, 53]. Taxonomic resolution at the species or strain level would allow better interpretation of these findings, though one possibility is that oral contamination secondary to grooming or other behaviors accounts for the over-abundance of these microbes in voided urine.
The biological significance of the single taxon, Burkholderia-Caballeronia-Paraburkholderia, over-represented in cystocentesis samples is unclear. This taxonomic group encompasses multiple genera with diverse functions that are associated with both environmental microbes, as well as human and animal pathogens [54]. Burkholderia has also been observed in healthy human urine [1, 16]. However, the incomplete taxonomic resolution of this ASV and the identification of a separate Burkholderia-Caballeronia-Paraburkholderia ASV as a potential contaminant in this dataset complicate interpretation of this finding.
Urine collected via cystocentesis also exhibited significantly different UM composition compared to voided urine using both Bray Curtis and Unweighted UniFrac measures of beta diversity, but not Weighted UniFrac. This finding suggests that when taking both phylogenetic relatedness and the evenness of organism distribution into account (Weighted UniFrac), voided and cystocentesis samples are more similar. However, the overall presence or absence of organisms was distinct between groups, even when accounting for phylogenetic relatedness (Unweighted UniFrac), and the overall distribution of organisms when not considering phylogenetic relatedness also differed (Bray Curtis). More pronounced changes between urine collection methods via Unweighted UniFrac, as compared to Weighted UniFrac, also occurs in humans when comparing voided to catheterized urine samples [13]. The low R2 values for beta diversity by collection method (less than 20% across analyses) indicate that other variables also drive variation between samples. Low R2 values have also been observed in human studies evaluating urine collection method [16]. However, the degree of variation that is clinically relevant is unknown, and a low R2 might still equate to a meaningful difference.
While we did not detect differences in UM alpha and beta diversity between male and female dogs in this study, the ability to detect differences is likely impacted by study sample size, reproductive status of the dogs, and urine collection technique. For instance, a significant difference in the canine UM was observed in one study using voided samples [24], but not in a separate study that analyzed only cystocentesis samples [22]. In the current study, differential abundance testing within voided urine samples did identify two species that differed by sex: Haemophilus (over-represented in males) and Schaalia canis (over-represented in females). Haemophilus has been well-documented in the male urethra, with various strains of Haemophilus shown to associate with urethritis in men [55,56,57,58], and Schaalia canis, formerly known as Actinomyces canis [59, 60], has been previously isolated from the canine vagina [61]. These findings support the notion that urethro-genital microbes can contaminate the UM of voided canine urine samples and contribute to differences between sexes.
The discovery that collection method introduces differences in the UM highlights the need for researchers to employ consistent sample collection methods within a UM study. However, it is important to note that the current study was not designed to evaluate which urine collection technique is superior or more accurately reflects the UM. Rather, the technique should be selected based on the biological question of interest. For instance, a study assessing the UM in dogs with urethral or prostatic disease may warrant the use of voided samples, whereas a study evaluating the UM from dogs with bladder or upper urinary tract disorders may deem cystocentesis or other collection methods most informative. Additionally, researchers should exercise caution when interpreting results across studies that use different urine collection methods.
Towards our secondary objective, we found that patterns of significance for alpha and beta diversity remained largely consistent across analyses using different minimum sequence read thresholds and normalization methods. A rarefaction curve demonstrated that 300 sequence reads, the threshold used for the primary analysis, captured the majority of ASV diversity in most samples. The three methods of data normalization produced only minor differences in P values or R2 values. The overall consistency between results using various read thresholds and normalization methods offers confidence when interpreting the major findings of this study. However, this study was not designed to evaluate the accuracy or relative superiority of specific normalization techniques, and we direct readers to other resources for more in-depth discussions of data normalization methods, considerations for their use, and their overall performance when evaluating microbial ecosystems [42, 62, 63].
This study has several limitations to consider. First, this population included a small sample size, with a total of only 19 dogs included overall (38 urine samples), but only 13 dogs (26 urine samples) included in the primary analysis after filtering for those with greater than 300 reads present in both paired samples. Additionally, only spayed or neutered dogs were included to mitigate potential confounding variables related to hormonal status. In women, both menopausal-status and menstruation impact UM composition [64, 65]. To date, no studies have evaluated the impact of spay or neuter status on the canine UM; future studies incorporating the impact of collection method on sexually intact dogs is warranted. Another limitation of the study was UM characterization by amplicon sequencing of the bacterial 16 S rRNA gene. While this technique is efficient for taxonomically characterizing microbial communities, this method cannot determine the viability of detected organisms, offers limited taxonomic resolution beyond the genus level, and does not assess the functional potential of organisms. Expanded quantitative urine culture would provide more insight into the viability of these microbial populations [3], and shotgun metagenomics are needed to better define the organisms and their functions, though the low biomass of urine samples poses a challenge with this technique [66]. Finally, only cystocentesis and voided urine were evaluated in the present study; larger prospective studies that also include urethral catheterization or cystoscopic-collected urine could be performed to expand upon these findings.
Conclusion
Canine urine collected via cystocentesis harbors different microbial communities than urine collected via midstream voiding from the same dog. Contaminant microbes from other anatomic sites likely contribute to the greater abundance of unique microbial organisms in voided urine, supporting the current consensus that the cystocentesis samples better reflect bladder urine. Results were consistent across different analytical methods employed. In sum, urine collection technique affects the composition of urinary microbial communities in dogs. When possible, sample collection methods should be standardized when investigating the canine UM, and when not possible, collection technique should be considered a covariate in the analyses.
Data Availability
Raw sequence data generated by and analyzed within this study are available through the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under the BioProject number PRJNA859856. Other data are included in the manuscript or additional files.
Abbreviations
- ASV:
-
Amplicon sequence variant
- BCS:
-
Body condition score
- IV:
-
Indicator value
- PCoA:
-
Principal coordinates analysis
- PERMANOVA:
-
Permutational multivariate analysis of variance
- UM:
-
Urinary microbiota
- USG:
-
Urine specific gravity
- UUF:
-
Unweighted UniFrac
- WUF:
-
Weighted UniFrac
References
Wolfe AJ, Toh E, Shibata N, Rong R, Kenton K, FitzGerald MP, et al. Evidence of uncultivated bacteria in the adult female bladder. J Clin Microbiol. 2012;50:1376–83.
Price TK, Dune T, Hilt EE, Thomas-White KJ, Kliethermes S, Brincat C, et al. The clinical urine culture: enhanced techniques improve detection of clinically relevant microorganisms. J Clin Microbiol. 2016;54:1216–22.
Hilt EE, McKinley K, Pearce MM, Rosenfeld AB, Zilliox MJ, Mueller ER, et al. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J Clin Microbiol. 2014;52:871–6.
Fouts DE, Pieper R, Szpakowski S, Pohl H, Knoblach S, Suh MJ, et al. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J Transl Med. 2012;10:174.
Price TK, Hilt EE, Thomas-White K, Mueller ER, Wolfe AJ, Brubaker L. The urobiome of continent adult women: a cross-sectional study. BJOG. 2020;127:193–201.
Pearce MM, Zilliox MJ, Thomas-White KJ, Richter HE, Nager CW, Visco AG et al. The female urinary microbiome in urgency urinary incontinence. Am J Obstet Gynecol. 2015;213:347.e1-347.e11.
Pearce MM, Hilt EE, Rosenfeld AB, Zilliox MJ, Thomas-White K, Fok C, et al. The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. MBio. 2014;5:1–12.
Zampini A, Nguyen AH, Rose E, Monga M, Miller AW. Defining dysbiosis in patients with Urolithiasis. Sci Rep. 2019;9:1–13.
Wu P, Zhang G, Zhao J, Chen J, Chen Y, Huang W, et al. Profiling the urinary microbiota in male patients with bladder cancer in China. Front Cell Infect Microbiol. 2018;8:1–10.
Hourigan SK, Zhu W, Wong SWW, Clemency NC, Provenzano M, Vilboux T, et al. Studying the urine microbiome in superficial bladder cancer: samples obtained by midstream voiding versus cystoscopy. BMC Urol. 2020;20:1–8.
Brubaker L, Putonti C, Dong Q, Wolfe AJ, February. https://doi.org/10.1007/s00335-021-09862-8.
Brubaker L, Gourdine J-PF, Siddiqui NY, Holland A, Halverson T, Limeria R, et al. Forming Consensus to Advance Urobiome Research. mSystems. 2021;6:e0137120.
Pohl H, Groah SL, Perez-Losada M, Ljungberg I, Sprague BM, Chandal N, et al. The urine microbiome of healthy men and women differs by urine collection method. Int Neurourol J. 2020;24:41–51.
Chen YB, Hochstedler B, Pham TT, Alvarez MA, Mueller ER, Wolfe AJ. The urethral microbiota: a missing link in the female urinary microbiota. J Urol. 2020;204:303–9.
Dong Q, Nelson DE, Toh E, Diao L, Gao X, Fortenberry JD, et al. The microbial communities in male first catch urine are highly similar to those in paired urethral swab specimens. PLoS ONE. 2011;6:1–5.
Hrbacek J, Morais D, Cermak P, Hanacek V, Zachoval R. Alpha–diversity and microbial community structure of the male urinary microbiota depend on urine sampling method. Sci Rep. 2021;11:23758.
Southworth E, Hochstedler B, Price T, Joyce C, Wolfe A, Mueller E. A cross-sectional pilot cohort study comparing standard urine collection to the Peezy midstream device for research studies involving women. Female Pelvic Med Reconstr Surg. 2019;25:e28–33.
Alford A, Furrow E, Borofsky M, Lulich J. Animal models of naturally occurring stone disease. Nat Rev Urol. 2020;17:691–705.
Ruetten H, Vezina CM. Relevance of dog as an animal model for urologic diseases. 1st edition. Elsevier Inc.; 2022.
Knapp DW, Ramos-Vara JA, Moore GE, Dhawan D, Bonney PL, Young KE. Urinary bladder cancer in dogs, a naturally occurring model for cancer biology and drug development. ILAR J. 2014;55:100–18.
Cook L, Byron J, Moore S. Urological sequelae to acute spinal cord injury in pet dogs: a natural disease model of neuropathic bladder dysfunction. Top Spinal Cord Inj Rehabil. 2019;25:205–13.
Burton EN, Cohn LA, Reinero CN, Rindt H, Moore SG, Ericsson AC. Characterization of the urinary microbiome in healthy dogs. PLoS ONE. 2017;12:e0177783.
Melgarejo T, Oakley BB, Krumbeck JA, Tang S, Krantz A, Linde A. Assessment of bacterial and fungal populations in urine from clinically healthy dogs using next-generation sequencing. J Vet Intern Med. 2021;35:1416–26.
Mrofchak R, Madden C, Evans MV, Hale VL. Evaluating extraction methods to study canine urine microbiota. PLoS ONE. 2021;16:e0253989.
Coffey EL, Gomez AM, Burton EN, Granick JL, Lulich JP, Furrow E. Characterization of the urogenital microbiome in miniature schnauzers with and without calcium oxalate urolithiasis. J Vet Intern Med. 2022;36:1341–52.
Mrofchak R, Madden C, Evans MV, Kisseberth WC, Dhawan D, Knapp DW, et al. Urine and fecal microbiota in a canine model of bladder cancer and comparison of canine and human urine microbiota. All Life. 2022;15:1245–63.
Weese JS, Blondeau JM, Boothe D, Breitschwerdt EB, Guardabassi L, Hillier A, et al. Antimicrobial use guidelines for treatment of urinary tract disease in dogs and cats: antimicrobial guidelines working group of the international society for companion animal infectious diseases. Vet Med Int. 2011;2011:263768.
Yu Y, Pieper R. Urinary Pellet Sample Preparation for Shotgun Proteomic Analysis of Microbial infection and host Pathogen interactions. Methods Mol Biol. 2015;1295:65–74.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108 SUPPL. 1:4516–22.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. 2011;17:10–2.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:590–6.
Miller AW, Dale C, Dearing MD. The induction of Oxalate Metabolism in vivo is more effective with functional Microbial Communities than with functional Microbial Species. mSystems. 2017;2:1–12.
Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:1–14.
Oksanen J, Blanchet G, Friendly M, Kindt R, Legendre P, McGlinn D et al. Vegan: Community Ecology Package. R package version 2.5-7. URL http//CRANRproject.org/package = vegan. 2020.
McMurdie PJ, Holmes S. Phyloseq: an R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE. 2013;8:e61217.
Dufrêne M, Legendre P. Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol Monogr. 1997;67:345–66.
Roberts DW, labdsv. Ordination and Multivariate Analysis for Ecology. R package version 1.3. 2007. URL http://CRAN.R-project.org/package=labdsv. 2019.
Sharma AK, Davison S, Pafco B, Clayton JB, Rothman JM, McLennan MR, et al. The primate gut mycobiome-bacteriome interface is impacted by environmental and subsistence factors. npj Biofilms Microbiomes. 2022;8:1–11.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1–21.
Lin H, Peddada S. Analysis of microbial compositions: a review of normalization and differential abundance analysis. npj Biofilms Microbiomes. 2020;6:60.
Hutchins RG, Vaden SL, Jacob ME, Harris TL, Bowles KD, Wood MW, et al. Vaginal microbiota of spayed dogs with or without recurrent urinary tract infections. J Vet Intern Med. 2014;28:300–4.
Rota A, Corrò M, Patuzzi I, Milani C, Masia S, Mastrorilli E, et al. Effect of sterilization on the canine vaginal microbiota: a pilot study. BMC Vet Res. 2020;16:1–10.
Lehtimäki J, Sinkko H, Hielm-Björkman A, Salmela E, Tiira K, Laatikainen T, et al. Skin microbiota and allergic symptoms associate with exposure to environmental microbes. Proc Natl Acad Sci U S A. 2018;115:4897–902.
Tang S, Prem A, Tjokrosurjo J, Sary M, Van Bel MA, Rodrigues-Hoffmann A, et al. The canine skin and ear microbiome: a comprehensive survey of pathogens implicated in canine skin and ear infections using a novel next-generation-sequencing-based assay. Vet Microbiol. 2020;247:108764.
García-Fonticoba R, Ferrer L, Francino O, Cuscó A. The microbiota of the surface, dermis and subcutaneous tissue of dog skin. Anim Microbiome. 2020;2:1–9.
Cuscó A, Belanger JM, Gershony L, Islas-Trejo A, Levy K, Medrano JF, et al. Individual signatures and environmental factors shape skin microbiota in healthy dogs. Microbiome. 2017;5:139.
Pilla R, Suchodolski JS. The gut microbiome of dogs and cats, and the influence of Diet. Vet Clin North Am - Small Anim Pract. 2021;51:605–21.
Middelbos IS, Vester Boler BM, Qu A, White BA, Swanson KS, Fahey GC. Phylogenetic characterization of Fecal Microbial Communities of Dogs Fed Diets with or without Supplemental Dietary Fiber using 454 pyrosequencing. PLoS ONE. 2010;5:e9768.
Ujvári B, Orbán B, Incze Z, Psáder R, Magyar T. Occurrence of Pasteurellaceae and Neisseriaceae bacteria in the pharyngeal and respiratory tract of dogs and cats - short communication. Acta Vet Hung. 2020;68:231–5.
Bell SE, Nash AK, Zanghi BM, Otto CM, Perry EB. An Assessment of the Stability of the canine oral Microbiota after Probiotic Administration in Healthy Dogs over Time. Front Vet Sci. 2020;7:1–8.
Dewhirst FE, Klein EA, Thompson EC, Blanton JM, Chen T, Milella L, et al. The canine oral microbiome. PLoS ONE. 2012;7:e36067.
Estrada-de los Santos P, Palmer M, Chávez-Ramírez B, Beukes C, Steenkamp ET, Briscoe L, et al. Whole genome analyses suggests that Burkholderia sensu lato contains two additional novel genera (Mycetohabitans gen. nov., and Trinickia gen. nov.): implications for the evolution of diazotrophy and nodulation in the Burkholderiaceae. Genes (Basel). 2018;9:389.
Frølund M, Wikström A, Lidbrink P, Al-Soud WA, Larsen N, Harder CB, et al. The bacterial microbiota in first-void urine from men with and without idiopathic urethritis. PLoS ONE. 2018;13:1–19.
Srinivasan S, Chambers LC, Tapia KA, Hoffman NG, Munch MM, Morgan JL, et al. Urethral microbiota in men: Association of Haemophilus influenzae and Mycoplasma penetrans with Nongonococcal Urethritis. Clin Infect Dis. 2021;73:e1684–93.
Vives A, da Silva GVM, Alonso-Tarrés C, Suarez JB, Palmisano F, Cosentino M. Haemophilus urethritis in males: a series of 30 cases. Rev Int Androl. 2021;19:160–3.
Horie K, Ito S, Hatazaki K, Yasuda M, Nakano M, Kawakami K, et al. Haemophilus quentini” in the urethra of men complaining of urethritis symptoms. J Infect Chemother. 2018;24:71–4.
Hoyles L, Falsen E, Foster G, Pascual C, Greko C, Collins MD. Actinomyces canis sp. nov., isolated from dogs. Int J Syst Evol Microbiol. 2000;50:1547–51.
Nouioui I, Carro L, García-López M, Meier-Kolthoff JP, Woyke T, Kyrpides NC, et al. Genome-based taxonomic classification of the phylum Actinobacteria. Front Microbiol. 2018;9:2007.
Schaalia canis. IFR M2289/98/2 is a microaerophile, mesophilic, Gram-positive animal pathogen that was isolated from dog vagina. BacDive doi 101093/nar/gkab961.
Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:1–18.
McMurdie PJ, Holmes S. Waste not, want not: why rarefying Microbiome Data is inadmissible. PLoS Comput Biol. 2014;10:e1003531.
Ammitzbøll N, Bau BPJ, Bundgaard-Nielsen C, Villadsen AB, Jensen AM, Leutscher PDC, et al. Pre- and postmenopausal women have different core urinary microbiota. Sci Rep. 2021;11:1–10.
Price TK, Wolff B, Halverson T, Limeira R, Brubaker L, Dong Q, et al. Temporal dynamics of the adult female lower urinary tract microbiota. MBio. 2020;11:1–14.
Moustafa A, Li W, Singh H, Moncera KJ, Torralba MG, Yu Y, et al. Microbial metagenome of urinary tract infection. Sci Rep. 2018;8:1–12.
Acknowledgements
Not applicable.
Funding
The work in this study was funded by the University of Minnesota’s College of Veterinary Medicine Resident and Graduate Student Research Grant. Dr. Coffey was supported by an institutional NIH T32 Comparative Medicine and Pathology Training Grant (University of Minnesota, T32 OD010993-15). Partial support for Dr. Furrow was provided by an NIH ORIP K01 Mentored Research Scientist Development Award (K01-OD019912).
Author information
Authors and Affiliations
Contributions
All authors participated in the conception and design of this study. EC recruited participants for enrollment and assisted with the acquisition of samples. EC, EF, AG, and AE served major roles in the analysis of data. EC, AG, AE, EB, JG, JL, and EF each assisted in data interpretation, manuscript revisions, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Study protocols were approved by the University of Minnesota Institutional Animal Care and Use Committee (protocol # 2005-38140 A). Written, informed consent was obtained prior to study enrollment, and owners were able to withdraw animals from the study at any time. All methods were carried out in accordance with relevant guidelines and regulations, including the ARRIVE guidelines for the reporting of animal experiments.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Coffey, E.L., Gomez, A.M., Ericsson, A.C. et al. The impact of urine collection method on canine urinary microbiota detection: a cross-sectional study. BMC Microbiol 23, 101 (2023). https://doi.org/10.1186/s12866-023-02815-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-023-02815-y