
Abstract
Background
Soil microbiome is an important part of the forest ecosystem and participates in forest ecological restoration and reconstruction. Niche differentiation with respect to resources is a prominent hypothesis to account for the maintenance of species diversity in forest ecosystems. Resource-based niche differentiation has driven ecological specialization. Plants influence soil microbial diversity and distribution by affecting the soil environment. However, with the change in plant population type, whether the distribution of soil microbes is random or follows an ecologically specialized manner remains to be further studied. We characterized the soil microbiome (bacteria and fungi) in different plant populations to assess the effects of phytophysiognomy on the distribution patterns of soil microbial communities in a temperate forest in China.
Results
Our results showed that the distribution of most soil microbes in different types of plant populations is not random but specialized in these temperate forests. The distribution patterns of bacteria and fungi were related to the composition of plant communities. Fungal species (32%) showed higher specialization than bacterial species (15%) for different types of plant populations. Light was the main driving factor of the fungal community, and soil physicochemical factors were the main driving factor of the bacterial community.
Conclusion
These findings suggest that ecological specialization is important in maintaining local diversity in soil microbial communities in this forest. Fungi are more specialized than bacteria in the face of changes in plant population types. Changes in plant community composition could have important effects on soil microbial communities by potentially influencing the stability and stress resistance of forest ecosystems.
Similar content being viewed by others

Introduction
Soil microbiome not only plays a critical role in regulating ecological processes relevant to nutrient cycling and carbon [1], but also produces strong positive feedback to promote plant regeneration and succession [2]. Soil microbial communities’ kind mutualistic dependent associations with soil and plants to enhance nutrient absorption [3]. The synergistic effect between the aboveground plant community and the underground microbial community contributes significantly to the restoration and stability of the ecosystem [4,5,6,7]. Mangan et al. (2010) reported that plant–soil feedback is an important mechanism that can maintain species diversity and explain patterns of tree-species relative abundance in forests [8]. Although soil microbes play a critical role in regulating ecological processes relevant to nutrient cycling and carbon in forest ecosystems, the distribution patterns of soil microbes with changes in plant populations remain to be further studied.
Niche differentiation is a prominent mechanism that facilitates the maintenance of species diversity in forest biomes [9,10,11]. One manifestation of resource-based niche differentiation consists of ecological specialization, such that different species are best suited to different habitats [9, 12,13,14,15,16]. If niche-relevant environmental conditions are spatially structured, then species distribution ought to be mirrored by the association between species and different habitats [16,17,18]. Harms et al. (2001) quantified the association of woody plants with topographic habitats, and their inference provides a good idea for assessing the contribution of ecological specialization to species coexistence [9]. Many researchers have since suggested that coexisting woody plants in forest ecosystems have distinct habitat preferences and emphasized the importance of ecological specialization for woody plant assemblage characteristics [9, 13, 19, 20]. Most of these studies have focused on woody plant populations [9, 18,19,20]. Nevertheless, the relative contribution of ecological specialization to the maintenance of diversity in soil microbial communities remains unknown.
Community structure, understory vegetation, and soil structure differ among different plant populations, which provide diverse habitats for the growth of soil microbes [21, 22]. Differences also exist between the life history and physiological traits of plants [23]. The differences among vegetation species create conditions for the specificity of soil environment and biological community [24]. Different forest types will lead to different components of litter, and the decomposition rate of plant litters will vary [25]. Moreover, soil nutrients, physicochemical properties, and light vary among different vegetation types, which affect the distribution of soil microbes. Many studies have shown that different plant species affect soil microbial diversity through root exudates and root properties [3, 26]. However, the role of plant population partitioning in soil microbe diversity maintenance remains poorly known.
Many studies have confirmed that the lifestyles of fungi and bacteria and their responses to environmental changes are different [27, 28]. Soil bacteria have stronger adaptability than fungi in the face of environmental changes [28,29,30,31,32]. For example, climate extremes-induced changes in plant communities had long-lasting associations with bacterial communities and strongly governed their recovery, but much less so for fungal communities [28]. However, whether bacteria and fungi have different distribution patterns in the face of plant population type changes is not well understood.
In this study, we hypothesized that ecological specialization of plant population types is important for structuring soil microbial communities in temperate mountain forest ecosystems and hence would be important for the maintenance of their local species diversity. To test these hypotheses, we characterized soil bacterial and fungal communities to determine their spatial distribution in 18 woody plant populations belonging to six community types in a temperate forest in China. We examined the distribution preferences of soil microbes (soil bacteria and fungi) at the community level (correlation network) and species level (torus-translation test). We also evaluated the influence of the environment on microbial community among different plant populations by variance partitioning. Results can improve understanding of the distribution patterns of fungi and bacteria in response to changes in plant population types in forest ecosystems.
Materials and methods
Site description
This study was conducted in Baiyun mountain in Luoyang City of Henan province, east China (33°38'–33°34' N, 111°48'–111°52' E, 1500 m above sea level). The total area is 168 km2, and the mean annual temperature is 18℃, with an average temperature of 0.2℃ in the coldest month (January) and 27.3℃ in the hottest month (July) [33]. The average annual precipitation is 1200 mm [34,35,36].
The forest canopy closure is 98.5% in Baiyun mountain. The dominant species in the forest are Quercus aliena var. acutiserrata, Forsythia suspensa, Pinus armandii, Quercus serrata var. brevipetiolata, Sorbus hupehensis, and Larix gmelinii [37].
Sampling design
Differences in soil microbial distribution under different communities were explored. Based on a comprehensive investigation, 18 communities belonging to six community types were selected in the Baiyun Mountain National Nature Reserve. The six community types are Quercus aliena var. acutiserrata community (QAV), Quercus serrata var. brevipetiolata community (QSV), Larix gmelinii community (LAG), Pinus armandii community (PIA), Forsythia suspensa community (FOS), and Sorbus hupehensis community (SOH) (Fig. S1). These communities are the main community types in this area. QAV and QSV belong to broad-leaved forests, LAG and PIA belong to coniferous forests, and FOS and SOH belong to shrub forests. Table S1 shows detailed information about the six communities. Three 20 m × 20 m quadrats were set in each type of community. All woody plants ≥ 1 cm diameter at breast height (DBH) in each 20 m × 20 m quadrat was identified, measured, and recorded.
In each 20 m × 20 m quadrat, topsoil (0–20 cm) was selected and three replicates were randomly sampled with a soil corer. After the visible stones, roots, and litter were removed, three samples were mixed to obtain one composite sample. Finally, 18 mixed soil samples were obtained. Each soil sample was divided into two parts: one part was immediately stored at − 80 °C until DNA extraction, and the other part at 4 °C until analysis of soil physicochemical properties.
DNA extraction, amplification, and high-throughput sequencing
Soil total DNA was extracted from 0.5 g of the freeze-dried soil samples by using FastDNA® Spin Kit for Soil based on the manufacturer’s instructions. The reaction mixture had a total volume of 20 μL, including 10 ng of template DNA, 2 μL of 2.5 mM dNTPs, 4 μL of 5 × TransStart FastPfu Buffer, 0.4 μL of TransStart FastPfu DNA Polymerase, 0.8 μL of 5 μM forward primer, 0.8 μL of 5 μM reverse primer, and finally ddH2O up to 20 μL. PCR conditions were 3 min at 95 °C, followed by 27 cycles of 30 s at 95 °C, 30 s at 55 °C, and 30 s at 72 °C; and a final extension at 72 °C for 10 min. PCR reactions were performed in triplicate. The hypervariable region V4 of the bacterial 16S rRNA [38] gene was amplified with primer pairs 515F(5ʹ-GTGCCAGCMGCCGCGGTAA-3ʹ) and 806R(5ʹ-GGACTACHVGGGTWTCTAAT-3ʹ) by an ABI GeneAmp® 9700 PCR thermocycler (ABI, CA, USA). The hypervariable region ITS1 of the fungal ITS gene [39] was amplified with primer pairs ITS1F (5ʹ-CTTGGTCATTTAGAGGAAGTAA-3ʹ) and ITS2R (5ʹ-GCTGCGTTCTTCATCGATGC-3ʹ) by an ABI GeneAmp® 9700 PCR thermocycler (ABI, CA, USA). The PCR products were separated on 2% agarose gels and purified using AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) [39]. All purified amplicons were sequenced (2 × 300) on Illumina Miseq platform of Majorbio Bio-Pharm Technology Co. Ltd (Shanghai, China) [39].
Sequencing of bacterial and fungal raw data yielded 14,455,502 and 12,614,520 reads, respectively. The raw gene sequencing reads were demultiplexed, quality filtered by Trimmomatic, and incorporated by Fast Length Adjustment of Short reads (FLASH, v1.2.11) with specific criteria. After the demultiplexed and quality control, the number of bacterial and fungal sequences were 7,320,500 and 6,584,900, respectively. Operational taxonomic units (OTUs) with 97% similarity cutoff [40] were clustered using UPARSE (version 7.1, http://drive5.com/uparse/) from bacteria and fungi. Chimeric sequences were identified and removed. The taxonomy of each OTU representative sequence was analyzed by RDP Classifier (http://rdp.cme.msu.edu/) against the 16S rRNA and ITS database (e.g. Silva SSU128) by using confidence threshold of 0.7 [41].
Environmental data
The total station was used to record the elevation of the plots. Slope, convex–concave, aspect, and mean elevation were calculated based on the elevation of the plots. Topographic factors were calculated using the methods of Harms et al.(2001) and Valencia et al.(2004) [9].
SLM9-UM-1.2 canopy analyzer (Delta-T Devices Co, Ltd.) was used to collect light environment data [42]. Light environment parameters include light transmittance (LT), scattered radiation (SR), total radiation (TR), canopy cover (CC), average leaf angle (ALA), and leaf area index (LAI).
Soil physicochemical properties evaluated include pH, soil moisture content (SWC), nitrogen (N), phosphorus (P), and soil organic matter (SOM) content [43,44,45,46]. Differences in environmental factors (topographic, light, soil) among different communities are shown in Figure S2.
Data analysis
Three microbial groups, namely, all species, core species, and dominant species, were constructed to understand the species information of different groups. We defined core species as those having relative abundances above 10% of all species [47,48,49], and dominant species as those having relative abundances above 0.5% of all species [50]. Core and dominant species have an important ecological role in microbiome assembly and ecosystem functions [51, 52].
Venn diagrams were plotted to show the number of OTUs that are unique to and shared among different communities and visualized using the VennDiagram package in R [53]. Rarefaction curves of the observed bacterial and fungal OTUs were calculated in each bacterial and fungal community (i.e. all 18 quadrats) by using the Specaccum command within the VEGAN package [54]. Percentage stacking diagrams were generated using R to show the relative abundance of dominant bacterial and fungal communities at genus and species levels [55]. The species composition of bacteria and fungi was analyzed by ordination using nonmetric multidimensional scaling (NMDS) with Bray Curtis dissimilarity, and the different types of communities were fitted as centroids onto the NMDS graph using the envfit function. NMDS was conducted using the metaMDS command within the VEGAN package [16, 54].
Network analysis was used to analyze the specificity of microbes to different plant populations. Network analysis data were visualized using Gephi software [56]. The network was compartmentalized into six modules of closely associated bacteria and fungi. The structure of the community–microbe network was evaluated using modularity index [54]. This parameter was calculated using the network-level function of the bipartite package [57].
Torus-translation test is currently the most used method for determining the association between species and habitat [19, 58, 59]. This test calculates the probability of the true distribution of a species in each habitat under the condition of random distribution and determines whether a species is significantly correlated with a certain type of habitat through probability analysis. In addition, the test considers the spatial autocorrelation of species distribution (Harms et al., 2001; for details) [9]. In the present study, torus-translation test was used to examine the associations between microbes (11,610 OTUs of bacteria and 4340 OTUs of fungi) and community in Baiyun mountain. Prior to data analysis, OTUs with relative abundances less than 0.01% were removed [48]. The associations of all species, core species, dominant species with community type (positive correlations, P ≤ 0.05) were analyzed by torus-translation test.
Variance partitioning was used to distinguish the results of various environmental factors on microbial communities (all species, core species, dominant species) [60]. Variations in species compositions in soil bacteria and fungi were partitioned into topographical factors (aspect, slop, elevation, and convex and concave), soil physicochemical properties (pH, P, N, SWC, and SOM), and light (LT, SR, TR, CC, LAI, and ALA) by using the Varpart function in the VEGAN package [54].
All statistical analyses were conducted in R 4.0.3.
Results
Change to microbiome composition among different phytophysiognomies
The Venn diagram showed that the OTU numbers of bacteria and fungi varied among different communities (Fig. 1). The highest number (8200) of OTUs of bacteria was found in the PIA community. The maximum (1844) number of OTUs of fungi was detected in LAG community.

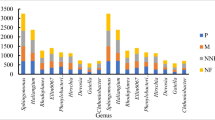
Venn diagrams showing the number of OTUs that were unique to and shared among different communities. Rarefaction curves of the observed OTU numbers of all the samples from the six types of communities after rarefied. Percentage stacking diagrams show the community composition of bacteria and fungi at genus and species levels. The top 30 genera and species were selected for abundance analysis. Three quadrats were set for each population, with 18 quadrats in 6 populations. The abscissa is the proportion of species in the sample, and the ordinate is the plot. Different colored columns represent different species, and the length of the columns represents the proportion of the size of the species. The abbreviations of species are shown in Supplementary Table S4
The rarefaction curve tended to flatten as the number of measured sequences increased (Fig. 1). At both genus and species levels, the diversity of fungal richness of the six plant populations was greater than that of bacteria (Fig. S3). The percentage stacking diagrams showed that the dominant species of bacteria at the genus and species levels were similar, but their relative abundance was different. The three most abundant genera were Udaeobacter (13.04%), Subgroup_2 (9.41%), and Bradyrhizobium (6.17%). The three most abundant species were Udaeobacter sp. (19.48%), Rokubacteriales sp. (13.96%), and Acidobacteria sp. (10.12%). The dominant species and relative abundance of fungi differed at the genus and species levels. The three most abundant genara were Russula (31.29%), Mortierella (19.94%), and Sebacina (11.17%). The three most abundant species were Sebacina sp. (13.66%), Russula vesca (9.43%), and Mortierella elongata (9.02%) (Fig. 1). The results of NMDS showed significant differences in the species composition of bacteria and fungi among different communities (all species of bacteria: P = 0.038; all species of fungi: P = 0.045) (Fig. 2).

NMDS analysis of species composition among the six types of communities. Different colored dots indicate different types of communities. The ellipse has a 95% confidence interval. PERMANOVA was used to evaluate intercommunity significance
Species specialization characteristics at community level
We detected six interconnected modules with different species compositions of fungi and bacteria among the communities (Fig. 3). The modularity index values were 0.15 for all bacterial species (Core: 0.12; Dominant: 0.17) and 0.32 for all fungal species (Core: 0.37; Dominant: 0.57) (Fig. 3).

Network analysis of OTU of bacteria and fungi in the six types of communities. The size of the node indicates the richness of the species. The color of the node indicates the distribution of species in different communities. A, B, C, D, E, and F were the Quercus aliena var. acutiserrata community, Quercus serrata var. brevipetiolata community, Larix gmelinii community, Pinus armandii community, Forsythia suspensa community, and Sorbus hupehensis community, respectively
Species specialization characteristics at species level
Based on torus-translation tests, positive associations with the six habitats were observed among 3811 out of the 11,610 (32.83%) and 3717 out of the 4340 (85.65%) examined bacterial and fungal species, respectively (P < 0.05). Most species were positively correlated with LAG community, with 1168 (1168/3811) bacterial species and 1051 (1051/3717) fungal species, accounting for one-third of the total positive correlation number (Fig. 4).

Bar diagrams of bacterial and fungal distribution at the OTU level. The bar diagrams show the percent of bacteria and fungi associated with the six types of communities. Association between microbe and community was tested by torus-translation random test (Torus-translation test, P ≤ 0.05 significance level)
The torus translation showed that 21.17% (3377/15950) of the species tended to be distributed in coniferous forests, while 13.77% (2197/15950) and 13.40% (2137/15950) of the species were distributed in broad-leaved forests and shrub forests, respectively. A total of 2.15%, 6.99%, 10.06%, 3.20%, 5.25%, and 5.19% bacteria OTUs showed positive associations (P < 0.05) with QAV, QSV, LAG, PIA, FOS, and SOH communities, respectively. A total of 15.55%, 10.62%, 24.22%, 18.13%, 11.59%, and 9.75% fungi OTUs showed positive associations (P < 0.05) with QAV, QSV, LAG, PIA, FOS, and SOH community, respectively. No species was negatively correlated with the communities (Fig. 4). In addition, some species (bacteria: 67.17%; fungi: 14.35%) were found to be neutral to all communities. Tables S2 and S3 show the detailed associations between species and communities.
Effect of environmental factors on soil microbial communities
Variance partitioning analysis showed that all environmental factors (topography, understory light availability, and soil physicochemical properties) explained 89.47% and 90.82% of the overall variation of bacteria and fungi, respectively (Fig. 5). Soil properties (28.69%) explained more variation in bacterial community than light (19.93%) and topography variables (23.77%). Light factors (37.08%) explained more variation in fungal community than topography (24.84%) and soil variables (30.32%) (Fig. 5).

Variance partitioning of the effects of soil, topography, and light on the species abundance of soil bacteria (first row) and soil fungi (second row). Numbers indicate the proportions of explained variation (adjusted R2 values). Values less than zero are not shown. Soil pH, soil water content, N, P, and soil organic matter. Topographical factors: elevation, slope, aspect, and convex–concave. Light: light transmittance, scattered radiation, total radiation, canopy cover, leaf area index, and average leaf angle
Discussion
In this study, the distribution pattern of most soil microbes in different types of plant populations is not random but specialized. Different microbial species show different plant population preferences. Fungal species showed higher specialization than bacterial species in different types of plant populations. The main environmental factors driving bacterial and fungal distribution vary among different types of plant populations in this temperate mountain forest. These findings suggest that ecological specialization is important in maintaining local diversity in soil microbial communities at local scales.
Not random but specialization
The characteristics of bacteria and fungi assemblages differed among different plant population types. The specificity of spatial distribution of soil microbes may be determined by abiotic environmental factors (e.g., topography, understory light availability, and soil physicochemical properties) and biological environmental factors (e.g. plant composition, and community structure) [61, 62]. Each plant population is characterized by particular vegetation communities, soil properties, and soil microbial community structure [63, 64]. In general, changes in the soil microbial community distribution reflect changing interactions between microbes and their environment [65, 66]. Plant communities influence microbial distribution through direct host-microbial interactions and rhizosphere effects [67] and indirect regulation of soil physicochemical properties [68]. Differences in canopy structure and litterfall and plant species composition among different communities affect understory light availability and soil physicochemical properties [25, 69, 70]. Different types of plant populations may also be the reason for the moderate modularity observed in the microbial–community networks (Fig. 3). Therefore, the distribution pattern of most soil microbes in different types of plant populations is not random but specialized in temperate forests.
Different plant population preferences
NMDS analysis showed that differences in plant populations could affect the community structure of soil microbes. Plant residues are the main source of nutrients for bacteria and fungi, and plant composition varies greatly among different plant communities, leading to significant microbial differences [71,72,73]. In the torus translation test, 85.65% (3717/4340) of the fungal species and 32.83% (3811/11610) of the bacterial species were associated with specific community types. The torus-translation test also showed that some of the species (bacteria: 67.17%; fungi: 14.35%) examined were not significantly related to any population. The lack of obvious ecological preferences in these species may be due to their wide niche width and high resistance to environmental change. Plant diversity [36], identity [74], and composition [75] also influence microbial distribution in forest ecosystems. Therefore, different microbial species show different plant population preferences in temperate forests.
In recent years, increasing reports have been reported on similar dominant soil bacterial groups in various ecosystems [76,77,78]. Fierer and Jackson (2006) [79] showed that the species composition of Acidobacteria, Actinobacteria, Proteobacteria, and Bacteroidetes dominated the bacterial community, and no significant change in different biologic communities was found. For example, Acidobacteria, Actinobacteria, and Proteobacteria account for more than 75% of the bacterial sequence in the soil of Changbai Mountain in northern China [78]. Proteobacteria, Acidobacteria, Actinobacteria and Verrucomicrobia are the dominant bacterial groups in Shennongjia, Southern China [80]. Proteobacteria, Acidobacteria and Verrucomicrobia contributed more than 60% of the soil bacterial sequence in the BCI 50 ha large plot [81]. In this study, Proteobacteria (27.89%), Acidobacteria (21.22%) and Verrucomicrobia (14.39%) accounted for more than 63% of the soil bacteria sequences that could be classified. This implies that Proteobacteria, Acidobacteria and Verrucomicrobia are likely the phylum principally reacting to plant population richness. As for soil fungi, Tedersoo et al. (2014) [82] studied the diversity and geographical distribution pattern of global soil fungi and showed that Basidiomycota and Ascomycota were the two most abundant groups in all ecosystems, but their relative proportions varied differently in all biotic communities. The soil fungal community of the BCI 50 ha plot was mainly composed of Basidiomycota and Ascomycota, with a relative abundance of 66% and 27%, respectively [81]. This study also showed that Ascomycota and Basidiomycota were the dominant groups of soil fungal communities, contributing 60.96% and 27.93%, respectively, accounting for 88% of all taxonomic fungal sequences. Hence, Ascomycota and Basidiomycota are likely the phylum principally reacting to plant population richness. Therefore, the dominant species of soil bacterial community and soil fungal community in different plant populations are similar, but their relative abundance is significantly different.
Our results showed that soil microbes preferred to be distributed in coniferous forests. Different components of litter among different forest types possibly affect the decomposition rate of litter, leading to differences in soil nutrients and properties, and thus affecting the distribution of microbes [25]. Soil nutrients have an important influence on plant-(above-ground) microbial interactions in forests, which may reinforce microbe-environment associations [13, 14]. Soil properties, especially organic matter and pH, are critical factors that govern the microbial assembly processes in forest ecosystems [3, 83,84,85]. In the present work, the contents of P, N, pH, and SWC in coniferous forest were higher than those in broad-leaved and shrub forests (Fig. S2). Therefore, coniferous forests may be the preferred habitat for many soil microbes in temperate regions. Our study demonstrates the importance of different types of plant populations in maintaining local diversity in soil microbial communities in temperate forests.
Fungi are more specialized than bacteria
Consistent with our prediction, fungal species showed higher specialization than bacterial species in different types of plant populations. The modularity index of fungi (32.00%) was higher than that of bacteria (15.00%). In addition, more fungi (85.65%) had specific preferences than bacteria (32.83%) with respect to different types of plant populations. In general, bacteria are more resilient than fungi in the face of environmental changes due to their relatively high intrinsic growth rates and unicellular nature [27]. Fungi are more closely related to plants roots than bacteria [3]. Plants can affect the survival of soil microbes by changing the input of root exudates [86, 87]. Fungi coevolve with host plants, forming mutualistic symbiosis during evolution [3]. Fungal communities are coupled with plant communities in forest ecosystems [3]. The plant–fungi coupling in a forest ecosystem may be due to the direct effects of the plant community through host–fungi specificity [67, 88], indirect effects through input of litter resources [86, 87], or plant-driven changes in soil physicochemical characteristics [68]. For example, abundant ectomycorrhizal trees (e.g., Larix gmelinii and Pinus armandii) could develop strong biotic interactions with ectomycorrhizal fungi [26]. Soil properties also can determine host–microbe and soil fungal communities interactions [89,90,91]. AM fungal communities, for example, may be driven more strongly by soil characteristics than host-symbiotic interactions [91]. Some autotrophic bacteria can obtain nutrients through chemosynthesis. Therefore, fungal community show more specialized than bacterial communities in the face of changes in plant population types.
Main drivers of fungal and bacterial communities are different in different types of plant populations
Ecological specialization is the process by which a species adapts to and persists in its living environment [92, 93]. Ecological specialization depends on species-specific relationships and local and contingent environmental constraints. This study found that main differences in influencing factors between bacterial and fungal communities in different types of plant populations. Light was the main driving factor of the fungal community, and soil physicochemical factors were the main driving factor of bacterial community. Light factors explained more variation in the fungal community than in the bacterial community. It has been reported that soil properties and nutrient content have a great influence on soil fungi [91, 94]. Soil properties and nutrients are important, but light is also the main factor affecting the distribution of soil fungi. Fungi are more sensitive to changes in light than bacteria [95]. Forest canopy is a crucial factor that affects the distribution of soil microbes [96, 97]. Different forest canopy structures will lead to different levels of understory light availability, which will lead to stronger specialization of fungi than bacteria [42]. Therefore, understory light availability may be one of the factors that contribute to the higher specificity of fungal community than bacterial community. The soil physicochemical factors explained more variations in the bacterial community than in the fungal community. As an important part of the soil ecosystem, soil bacteria play an active role in regulating soil nutrient cycling and soil carbon and nitrogen cycling [28]. Soil organic matter affects the survival of soil microbes by affecting soil physicochemical properties and soil matrix composition [61]. Forest litter provides high-quality substrate, which promotes the growth and reproduction of bacteria adapted to high-nutrient environment [61, 98]. The distribution of fungi is more influenced by plant community structure and composition than that of bacteria [99]. In this study, the influence of canopy structure on fungi was greater than that of bacteria. Hence, the main environmental factors driving bacterial and fungal distribution vary among different types of plant populations.
Conclusion and implications
Our findings are of great significance for understanding how complex soil microbial communities respond to changes in plant populations. Fungal species showed higher specialization than bacterial species in different types of plant populations. Changes in plant population types could have important effects on soil microbial communities by potentially influencing the stability and stress resistance of forest ecosystems. These findings underscore that in sustainable forest management, diverse plant populations should be maintained, since plant populations variability will promote soil microbial diversity. Our study also has its limitations. We did not identify mycorrhizal fungi species among the species detected. However, many fungi show host specificity and are thought to be closely related to plant communities. Therefore, studying the relationship between mycorrhizal fungi and host is the focus of future research.
Availability of data and materials
The raw reads of sequencing data is available at NCBI BioProject SRA database under the accession number PRJNA785719.
References
Wang KB, Zhang YW, Tang ZS, Shangguan ZP, Chang F, Jia FA, et al. Effects of grassland afforestation on structure and function of soil bacterial and fungal communities. Sci Total Environ. 2019;676:396–406.
Manuel EL, Manuel D. Plant diversity and soil stoichiometry regulates the changes in multifunctionality during pine temperate forest secondary succession. Sci Total Environ. 2019;697:134204.
Liu L, Zhu K, Krause SMB, Li S, Wang X, Zhang Z, et al. Changes in assembly processes of soil microbial communities during secondary succession in two subtropical forests. Soil Biol Biochem. 2021;154:108144.
Meisner A, De Deyn GB, de Boer W, van der Putten WH. Soil biotic legacy effects of extreme weather events influence plant invasiveness. P Natl Acad Sci USA. 2013;110(24):9835–8.
Kröel-Dulay G, Ransijn J, Schmidt IK, Beier C, De Angelis P, de Dato G, et al. Increased sensitivity to climate change in disturbed ecosystems. Nat Commun. 2015;6(1):1–7.
Kaisermann A, de Vries FT, Griffiths RI, Bardgett RD. Legacy effects of drought on plant-soil feedbacks and plant-plant interactions. New Phytol. 2017;215(4):1413–24.
Geisen S, Heinen R, Andreou E, van Lent T, Ten HFC, Thakur MP. Contrasting effects of soil microbial interactions on growth-defence relationships between early- and mid-successional plant communities. New Phytol. 2021;233(3):1345–57.
Mangan SA, Schnitzer SA, Herre EA, Mack KML, Valencia MC, Sanchez EI, et al. Negative plant-soil feedback predicts tree-species relative abundance in a tropical forest. Nature. 2010;466(7307):710–52.
Kyle EH, Richard C, Stephen PH, Robin BF. Habitat Associations of Trees and Shrubs in a 50-Ha Neotropical Forest Plot. J Ecol. 2001;89(6):947–59.
Nathan JBK, Renato V, David DA. Functional Traits and Niche-Based Tree Community Assembly in an Amazonian Forest. Science. 2008;322(5901):580–2.
Benjamin B. Hypervolume concepts in niche- and trait-based ecology. Ecography. 2018;41(9):1441–55.
Jonathan S. Plant coexistence and the niche. Trends Ecol Evol. 2004;19(11):605–11.
Griffin EA, Traw MB, Morin PJ, Pruitt JN, Wright SJ, Carson WP. Foliar bacteria and soil fertility mediate seedling performance: a new and cryptic dimension of niche differentiation. Ecology. 2016;97(11):2998–3008.
Griffin EA, Wright SJ, Morin PJ, Carson WP. Pervasive interactions between foliar microbes and soil nutrients mediate leaf production and herbivore damage in a tropical forest. New Phytol. 2017;216(1):99–112.
Chen Y, Yuan Z, Bi S, Wang X, Ye Y, Svenning J. Macrofungal species distributions depend on habitat partitioning of topography, light, and vegetation in a temperate mountain forest. Sci Rep-UK. 2018;8(1):1–13.
Chen Y, Shao Y, Xi J, Yuan Z, Ye Y, Wang T. Community Preferences of Woody Plant Species in a Heterogeneous Temperate Forest China. Front Ecol Evol. 2020;8:165.
Jens-Christian S. Microhabitat Specialization in a Species-Rich Palm Community in Amazonian Ecuador. J Ecol. 1999;87(1):55–65.
Guo Y, Li D, Wang B, He Y, Xiang W, Jiang Y, et al. Composition and spatio-temporal dynamics of litter fall in a northern tropical karst seasonal rainforest in Nonggang, Guangxi, southern China. Biodiversity Sci. 2017;25(3):265.
Comita LS, Condit R, Hubbell SP. Developmental Changes in Habitat Associations of Tropical Trees. J Ecol. 2007;95(3):482–92.
Lai J, Mi X, Ren H, Ma K. Species-Habitat Associations Change in a Subtropical Forest of China. J Veg Sci. 2009;20(3):415–23.
Yuan Z, Gazol A, Wang X, Xing D, Lin F, Bai X, et al. What happens below the canopy? Direct and indirect influences of the dominant species on forest vertical layers. Oikos. 2012;121(7):1145–53.
Jia H, Chen Y, Wang X, Li P, Yuan Z, Ye Y. The Relationships among Topographically-driven Habitats, Dominant Species and Vertical Layers in Temperate Forest in China. Russ J Ecol. 2019;50(2):172–86.
Valerie TE. Plant Traits That Influence Ecosystem Processes Vary Independently among Species. Ecology. 2004;85(8):2215–29.
De Deyn GB, Cornelissen JHC, Bardgett RD. Plant functional traits and soil carbon sequestration in contrasting biomes. Ecol Lett. 2008;11(5):516–31.
Sun HZ. Microbial community Characteristics of different forest types and their relationship with environmental factors in permafrost region of Daxing’anling. China: Harbin Normal University; 2019.
Su L, Cheng A, Lin Y, Fu W, Zheng P. Investigation on mycorrhizae of forest trees in natural Reserve of mount tianmu. J Zhejiang For Coll. 1992;9:263–76.
Powell JR, Karunaratne S, Campbell CD, Yao H, Robinson L, Singh BK. Deterministic processes vary during community assembly for ecologically dissimilar taxa. Nat Commun. 2015;6(1):1–10.
Franciska TDV, Rob IG, Mark B, Hayley C, Mariangela G, Hyun SG, et al. Soil bacterial networks are less stable under drought than fungal networks. Nat Commun. 2018;9(1):1–12.
Bapiri A, Bååth E, Rousk J. Drying-rewetting cycles affect fungal and bacterial growth differently in an arable soil. Microb Ecol. 2010;60(2):419–28.
Franciska TDV, Mira EL, Lisa B, Matthew AB, Søren C, Heikki MS, et al. Land use alters the resistance and resilience of soil food webs to drought. Nat Clim Change. 2012;2(4):276–80.
Barnard RL, Osborne CA, Firestone MK. Responses of soil bacterial and fungal communities to extreme desiccation and rewetting. ISME J. 2013;7(11):2229–41.
de Vries FT, Shade A. Controls on soil microbial community stability under climate change. Front Microbiol. 2013;4:265.
Bi H, Wang B, Yang H, Feng J, Zheng J, Gao X. Study on the dynamic change of the tree layer biomass of the community Quercus aliena var. acutiserrata-Pinus armandii of Baiyunshan Mountain in Henan Province. J Henan Agric Univ. 2014;48(06):736–40.
Lin B. Overview of Baiyun Mountain National Forest Park. Henan forestry. 1999;02:40.
Xu C, Chen Z, Hao C, Ding X. Research on the correlation between plant species diversity and its main environmental factors of Mt. Baiyunshan in the transitional region from warm temperate zone to subtropical zone. Ecol Enviro Sci. 2014;23(03):371–6.
Chen Y, Guo L, Yao C, Wei B, Yuan Z, Ye Y, et al. Community characteristics of a deciduous broad-leaved forest in a temperatesubtropical ecological transition zone: Analyses of a 5-hm2 forest dynamics plot in Baiyunshan Nature Reserve. Henan Provinc Acta Ecol Sin. 2017;37(17):5602–11.
Li L, Yang H, Wang J, Qin Y, Fu Q. Species Structure and Diversity of Plant Community of Luhua Valley in Baiyun Mountain of Henan Province. J Hunan Agric Univ. 2017;10:44–7.
Zhao A, Zhang J, Zhang S, Jiao Z. Soil bacterial diversity in the Baotianman deciduous broad-leaved forest. Biodiversity Sci. 2015;23(5):649–57.
Zhang N, Li Y, Wubet T, Bruelheide H, Liang Y, Purahong W, et al. Tree species richness and fungi in freshly fallen leaf litter: Unique patterns of fungal species composition and their implications for enzymatic decomposition. Soil Biol Biochem. 2018;127:120–6.
Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, et al. Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol. 2013;22(21):5271–7.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics (Oxford, England). 2011;27(21):2957–63.
Ma H, Wen Y, Han Y, Li S. Correlations between Light Distribution of the Crown Layer and Natural Regeneration of Fraxinus mandshurica. J Northwest Forestry Univ. 2021;36(02):97–101.
Lu N, Xu X, Wang P, Zhang P, Ji B, Wang X. Succession in arbuscular mycorrhizal fungi can be attributed to a chronosequence of Cunninghamia lanceolata. Sci Rep-UK. 2019;9(1):1–12.
Zhang J, He P, Liu Y. Du Wei, Jing H, Nie C: Soil properties and microbial abundance explain variations in N2O fluxes from temperate steppe soil treated with nitrogen and water in Inner Mongolia. China Appl Soil Ecol. 2021;165:103984.
Yang G, Ma Y, Jiang B, Ma H, Li Y. Analysis of the bacterial community and diversity in tea plantation soil via 16S rDNA sequencing. Acta Ecol Sin. 2019;39(22):8452–61.
Sui X, Zhang R, Frey B, Yang L, Li M, Ni H. Land use change effects on diversity of soil bacterial, Acidobacterial and fungal communities in wetlands of the Sanjiang Plain, northeastern China. Sci Rep-UK. 2019;9(1):1–14.
Magurran AE, Henderson PA. Explaining the excess of rare species in natural species abundance distributions. Nature. 2003;422(6933):714–6.
Jiao S, Yang Y, Xu Y, Zhang J, Lu Y. Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 2020;14(1):202–16.
Mo Y, Peng F, Gao X, Xiao P, Logares R, Jeppesen E, et al. Low shifts in salinity determined assembly processes and network stability of microeukaryotic plankton communities in a subtropical urban reservoir. Microbiome. 2021;9(1):1–17.
Xiong C, Zhu Y, Wang J, Singh B, Han L, Shen J, Li P, Wang G, Wu C, Ge A, et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2020;229(2):1091–104.
Banerjee S, Schlaeppi K, van der Heijden MGA. Keystone taxa as drivers of microbiome structure and functioning. Nat Rev Microbiol. 2018;16(9):567–76.
Manuel D, Angela MO, Tess EB, Alberto B, David JE, Richard DB, et al. A global atlas of the dominant bacteria found in soil. Science. 2018;359(6373):320–5.
Chen H, Boutros P. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12(1):1–7.
Olesen JM, Bascompte J, Dupont YL, Jordano P. The modularity of pollination networks. P Natl Acad Sci USA. 2007;104(50):19891–6.
Zhang H, Feng J, Chen S, Li B, Sekar R, Zhao Z, et al. Disentangling the Drivers of Diversity and Distribution of Fungal Community Composition in Wastewater Treatment Plants Across Spatial Scales. Front Microbiol. 2018;9:1291.
Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. Icwsm. 2009;3(1):361–2.
Dormann CF, Gruber B, Fründ J. Introducing the bipartite package: analysing ecological networks. R News. 2008;8:1609–3631.
Saara JD, Kalan I, Reuben N, Kyle EH, David FRPB. Liana Habitat Associations and Community Structure in a Bornean Lowland Tropical Forest. Plant Ecol. 2006;186(2):203–16.
Toshihiro Y, Akemi T, Akira I, Takuo Y, Tatsuhiro O, Mamoru K, et al. Habitat associations of Sterculiaceae trees in a Bornean rain forest plot. J Veg Sci. 2006;17(5):559–66.
Legendre P, Mi X, Ren H, Ma K, Yu M, Sun I, et al. Partitioning beta diversity in a subtropical broad-leaved forest of China. Ecology. 2009;90(3):663–74.
Wang P, Ren B, Yi C, Liu H. Relationship between vertical variation of soil physical and chemical properties and environmental factors in Jiaozishan Nature Reserve. J Mt Sci-Engl. 2013;31(4):456–63.
Deng J, Zhu W, Zhou Y, Yin Y, Bai X, Zhang H, et al. Effects of different land use patterns on the soil microbial community diversity in montane region of eastern Liaoning Province. China Chin J Appl Ecol. 2018;29(7):2269–76.
Uriarte M, Turner BL, Thompson J, Zimmerman JK. Linking spatial patterns of leaf litterfall and soil nutrients in a tropical forest: a neighborhood approach. Ecol Appl. 2015;25(7):2022–34.
Stephanie AY, Mona NH. Soil bacteria and archaea change rapidly in the first century of Fennoscandian boreal forest development. Soil Biol Biochem. 2017;114:160–7.
Harris JA. Measurements of the soil microbial community for estimating the success of restoration. Eur J Soil Sci. 2003;54(4):801–8.
Hansel CM, Fendorf S, Jardine PM, Francis CA. Changes in bacterial and archaeal community structure and functional diversity along a geochemically variable soil profile. Appl Environ Microb. 2008;74(5):1620–33.
Martinez-Garcia LB, Richardson SJ, Tylianakis JM, Peltzer DA, Dickie IA. Host identity is a dominant driver of mycorrhizal fungal community composition during ecosystem development. New Phytol. 2015;205(4):1565–76.
Donald RZ, William EH, David CW, Aaron DP, David T. Plant Diversity, Soil Microbial Communities, and Ecosystem Function: Are There Any Links? Ecology. 2003;84(8):2042–50.
Stark SC, Enquist BJ, Saleska SR, Leitold V, Schietti J, Longo M, et al. Linking canopy leaf area and light environments with tree size distributions to explain Amazon forest demography. Ecol Lett. 2015;18(7):636–45.
Liu Z, Wang Y, Liu Y, Tian A, Wang Y, Zuo H. Spatiotemporal variation and scale effect of canopy leaf area index of larch plantation on a slope of the semi-humid Liupan Mountains, Ningxia. China Chin J Plant Ecol. 2017;41(7):749–60.
Rodrigo K, Luiz UH, Sandro S. Colonisation of low- and high-quality detritus by benthic macroinvertebrates during leaf breakdown in a subtropical stream. Limnologica. 2014;45:61–8.
Wang J, Liu L, Wang X, Chen Y. The interaction between abiotic photodegradation and microbial decomposition under ultraviolet radiation. Global Change Biol. 2015;21(5):2095–104.
Joelle S, Enrico M, Trent N. Feed Your Friends: Do Plant Exudates Shape the Root Microbiome? Trends Plant Sci. 2018;23(1):25–41.
Gao C, Zhang Y, Shi N, Zheng Y, Chen L, Wubet T, et al. Community assembly of ectomycorrhizal fungi along a subtropical secondary forest succession. New Phytol. 2015;205(2):771–85.
Prober SM, Leff JW, Bates ST, Borer ET, Firn J, Harpole WS, et al. Plant diversity predicts beta but not alpha diversity of soil microbes across grasslands worldwide. Ecol Lett. 2015;18(1):85–95.
Fierer N, Lennon JT. The generation and maintenance of diversity in microbial communities. (Special Issue: Biodiversity). Am J Bot. 2011;98(3):439.
Wang J, Soininen J, Zhang Y, Wang B, Yang X, Shen J. Contrasting patterns in elevational diversity between microorganisms and macroorganisms. J Biogeogr. 2011;38(3):595–603.
Shen C, Xiong J, Zhang H, Feng Y, Lin X, Li X, et al. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol Biochem. 2013;57:204–11.
Fierer N, Jackson RB. From the Cover: The diversity and biogeography of soil bacterial communities. P Natl Acad Sci USA. 2006;103(3):626–31.
Zhang Y, Cong J, Lu H, Li G, Xue Y, Deng Y, et al. Soil bacterial diversity patterns and drivers along an elevational gradient on Shennongjia Mountain. China Microb Biotechnol. 2015;8(4):739–46.
Barberán A, McGuire KL, Wolf JA, Jones FA, Wright SJ, Turner BL, et al. Relating belowground microbial composition to the taxonomic, phylogenetic, and functional trait distributions of trees in a tropical forest. Ecol Lett. 2015;18(12):1397–405.
Leho T, Mohammad B, Sergei P, Urmas K, Nourou SY, Ravi W, et al. Global diversity and geography of soil fungi. Science. 2014;346(6213):1–11.
Dini-Andreote F, Stegen JC, van Elsas JD, Salles JF. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. P Natl Acad Sci USA. 2015;112(11):E1326–32.
Tripathi BM, Stegen JC, Kim M, Dong K, Adams JM, Lee YK. Soil pH mediates the balance between stochastic and deterministic assembly of bacteria. ISME J. 2018;12(4):1072–83.
Shao P, Liang C, Rubert-Nason K, Li X, Xie H, Bao X. Secondary successional forests undergo tightly-coupled changes in soil microbial community structure and soil organic matter. Soil Biol Biochem. 2019;128:56–65.
Reynolds HL, Packer A, Bever JD, Clay K. Grassroots ecology: Plant-microbe-soil interactions as drivers of plant community structure and dynamics. Ecology. 2003;84(9):2281–91.
David AW, Lawrence RW, Richard DB. Ecosystem Properties and Forest Decline in Contrasting Long-Term Chronosequences. Science. 2004;305(5683):509–13.
Griffin EA, Harrison JG, Kembel SW, Carrell AA, Wright SJ, Carson WP. Plant host identity and soil macronutrients explain little variation in sapling endophyte community composition: Is disturbance an alternative explanation? J Ecol. 2019;107(4):1876–89.
Christian LL, Michael SS, Mark AB, Noah F. The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol Biochem. 2008;40(9):2407–15.
Francesco V, Krishnapillai S, Emilio LG, Roberta M, Sheridan LW, Matteo L. Trichoderma –plant–pathogen interactions. Soil Biol Biochem. 2008;40(1):1–10.
Schappe T, Albornoz FE, Turner BL, Neat A, Condit R, Jones FA. The role of soil chemistry and plant neighbourhoods in structuring fungal communities in three Panamanian rainforests. J Ecol. 2017;105(3):569–79.
Poisot T, Bever JD, Nemri A, Thrall PH, Hochberg ME. A conceptual framework for the evolution of ecological specialisation. Ecol Lett. 2011;14(9):841–51.
Xi J, Shao Y, Li Z, Zhao P, Ye Y, Li W, et al. Distribution of Woody Plant Species Among Different Disturbance Regimes of Forests in a Temperate Deciduous Broad-Leaved Forest. Front Plant Sci. 2021;12:618524.
Geisianny AMM, Helson MMDV. Soil Yeast Communities in Revegetated Post-Mining and Adjacent Native Areas in Central Brazil. Microorganisms. 2020;8(8):1116.
Griffin EA, Carson WP. The Ecology and Natural History of Foliar Bacteria with a Focus on Tropical Forests and Agroecosystems. Bot Rev. 2015;81(2):105–49.
Harrison JG, Forister ML, Parchman TL, Koch GW. Vertical stratification of the foliar fungal community in the world’s tallest trees. Am J Bot. 2016;103(12):2087–95.
Stone BWG, Jackson CR. Canopy position is a stronger determinant of bacterial community composition and diversity than environmental disturbance in the phyllosphere. Fems Microbiol Ecol. 2019;95(4):fiz032.
David AW, Marie-Charlotte N, Olle Z, Christiane G. Determinants of litter mixing effects in a Swedish boreal forest. Soil Biol Biochem. 2003;35(6):827–35.
Dassen S, Cortois R, Martens H, de Hollander M, Kowalchuk GA, van der Putten WH, et al. Differential responses of soil bacteria, fungi, archaea and protists to plant species richness and plant functional group identity. Mol Ecol. 2017;26(15):4085–98.
Acknowledgements
We thank Baotianman Forest Ecosystem Research Station for their help.
Funding
This work was supported by the China Postdoctoral Science Foundation [2021M693400]; Young Talents project funded by Henan Agricultural University [111/30500744]; and Youth Foundation of Natural Science Foundation of Henan Province [212300410153].
Author information
Authors and Affiliations
Contributions
Yun Chen and Jingjing Xi designed the study. Man Xiao, Senlin Wang, Zhiliang Yuan, Yizhen Shao and Fengqin Liu performed the field experiments and conducted fieldwork. Jingjing Xi, Senlin Wang and Wenju Chen conducted the laboratory work. Yun Chen and Jingjing Xi analyzed the data. Jingjing Xi wrote the manuscript. Yun Chen and Jingjing Xi contributed equally to this work. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Sampling sites and plot division. Figure S2. Environmental factors of different communities. Figure S3. Rarefaction curves of bacteria and fungi. Table S1. Dominant species of different communities. Table S2. Significant associations of bacteria with different communities based on Torus test. Table S3. Significant associations of fungi with different communities based on Torus test. Table S4. Abbreviations for bacteria and fungi.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, Y., Xi, J., Xiao, M. et al. Soil fungal communities show more specificity than bacteria for plant species composition in a temperate forest in China. BMC Microbiol 22, 208 (2022). https://doi.org/10.1186/s12866-022-02591-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-022-02591-1