
Abstract
Background
Dextran sulfate sodium (DSS) replicates ulcerative colitis (UC)-like colitis in murine models. However, the microbial characteristics of DSS-triggered colitis require further clarification. To analyze the changes in gut microbiota associated with DSS-induced acute and chronic colitis.
Methods
Acute colitis was induced in mice by administering 3% DSS for 1 week in the drinking water, and chronic colitis was induced by supplementing drinking water with 2.5% DSS every other week for 5 weeks. Control groups received the same drinking water without DSS supplementation. The histopathological score and length of the colons, and disease activity index (DAI) were evaluated to confirm the presence of experimental colitis. Intestinal microbiota was profiled by 16S rDNA sequencing of cecal content.
Results
Mice with both acute and chronic DSS-triggered colitis had significantly higher DAI and colon histopathological scores in contrast to the control groups (P < 0.0001, P < 0.0001), and the colon was remarkably shortened (P < 0.0001, P < 0.0001). The gut microbiota α-diversity was partly downregulated in both acute and chronic colitis groups in contrast to their respective control groups (Pielou index P = 0.0022, P = 0.0649; Shannon index P = 0.0022, P = 0.0931). The reduction in the Pielou and Shannon indices were more obvious in mice with acute colitis (P = 0.0022, P = 0.0043). The relative abundance of Bacteroides and Turicibacter was increased (all P < 0.05), while that of Lachnospiraceae, Ruminococcaceae, Ruminiclostridium, Rikenella, Alistipes, Alloprevotella, and Butyricicoccus was significantly decreased after acute DSS induction (all P < 0.05). The relative abundance of Bacteroides, Akkermansia, Helicobacter, Parabacteroides, Erysipelatoclostridium, Turicibacter and Romboutsia was also markedly increased (all P < 0.05), and that of Lachnospiraceae_NK4A136_group, Alistipes, Enterorhabdus, Prevotellaceae_UCG-001, Butyricicoccus, Ruminiclostridium_6, Muribaculum, Ruminococcaceae_NK4A214_group, Family_XIII_UCG-001 and Flavonifractor was significantly decreased after chronic DSS induction (all P < 0.05).
Conclusion
DSS-induced acute and chronic colitis demonstrated similar symptoms and histopathological changes. The changes in the gut microbiota of the acute colitis model were closer to that observed in UC. The acute colitis model had greater abundance of SCFAs-producing bacteria and lower α-diversity compared to the chronic colitis model.
Similar content being viewed by others


Background
Inflammatory bowel disease (IBD) comprises of Crohn’s disease (CD) and ulcerative colitis (UC). The hallmark of CD is chronic colorectal inflammation, which manifests as diarrhea, bloody mucus stool, abdominal pain and other symptoms that severely affect patients’ quality of life. The pathogenesis of UC is complex, and involves genetic, immunological and microbiological factors.
The dextran sodium sulfate (DSS)-stimulated colitis model mimics the pathological damage and symptoms of human UC [1], and is routinely used for studying the pathogenesis and pharmacodynamics of UC [2]. DSS is a water-soluble negatively-charged sulfated polysaccharide that can erode the intestinal mucosa [3, 4]. DSS increases colon permeability by destroying interepithelial cell tight junctions, reducing mucin levels while simultaneously altering the resident microbiota [5, 6]. A compromised intestinal barrier allows penetration of harmful bacterial from the intestinal lumen into the systemic circulation, thereby triggering an inflammatory response. The efficacy of colitis induction depends on the concentration of DSS (typically 1–5%), dosage regimen (long- or short-term), animal strains (BALB/c and C57BL/6 mice are more susceptible), and feeding conditions [7]. With the development of 16S rDNA sequencing, the DSS-induced colitis model been proven to be particularly well-suited for analyzing colitis-associated microbial changes. DSS-induced acute and chronic colitis mimic histopathological damages associated with UC. Intermittent administration of different concentrations of DSS over varying durations result in alternating active and remission phases, which is reflective of disease progression in UC patients. In order to establish acute and chronic models of colitis in mice, either 3% or 2.5% DSS is added to the drinking water for varying durations [8, 9].
Short chain fatty acids (SCFAs) are organic fatty acids with aliphatic tails of six or fewer carbons, including acetate, propionate, butyrate, valproate and hexanoate, that are mainly produced by anaerobic microorganisms while fermenting indigestible carbohydrates. SCFAs inhibit the production of inflammatory cytokines in human intestinal epithelial cells in vitro, which promotes intestinal mucosal repair by mitigating inflammation [10]. In vitro studies have shown that SCFAs also exert an anti-tumor effect by reducing the secretion of tumor necrosis factor in intestinal epithelial cells [11]. In addition, SCFAs (especially butyrate) regulate gut microbiota by modulating the intestinal lumen pH to levels that are conducive to the growth of SCFAs-producing bacteria [12]. Acetate, propionate and butyrate are the most abundant SCFAs produced during anaerobic fermentation of dietary fibers in the intestine, and account for more than 85% of all SCFAs. The relative proportion of the three is about 3:1:1 [13], and affected by the host genotype, intestinal microbial composition and diet. Anaerobic bacteria in the colon play an important role in fermentation. The main SCFAs producing-bacteria in the human gut belong to the phylum Firmicutes, in particular Faecalibacterium prausnitzii and Clostridium leptum of family Ruminococcaceae, Eubacterium rectale and Roseburia spp. of Lachnospiraceae, as well as Butyricicoccus, Rikenella and Alistipes [14, 15]. In addition, the mucin-degrading bacteria Akkermansia muciniphila (phylum Verrucomicrobia) produces both propionate and acetate [16].
Consistent with our previous studies [17,18,19,20], we found that DSS-triggered acute and chronic colitis were similar in terms of disease activity, colon shortening and histopathological changes in the colon, but differed remarkably in terms of gut microbiota profiles. Given that the initiation and progression of UC centers heavily on gut microbiota proportions, the therapeutic potential of fecal microbiota transplantation is increasingly being assessed in UC animal models. The selection of appropriate colitis mouse models vis-à-vis the gut microflora has become particularly important. In this study, we analyzed the changes in gut microbiota associated with DSS-triggered acute and chronic colitis in a mouse model in order to provide a reference basis for selecting an appropriate model.
Results
Acute and chronic colitis were successfully induced by DSS exposure
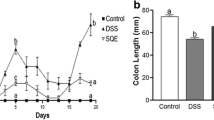
Mice exposed to DSS of varying concentrations and doses successfully developed acute and chronic colitis [8]. Based on preliminary results, we administered 3% DSS for 1 week to stimulate acute colitis and 2.5% DSS intermittently for 5 weeks to induce chronic colitis. As shown in Fig. 1A and B, the DAI scores of mice increased significantly after 1 week of administering 3% DSS compared to the untreated controls (P < 0.0001). However, the DAI scores fluctuated slightly after 14 days during chronic colitis modeling, corresponding to transient weight loss without change in stool characteristics. This can be attributed to intermittent DSS administration over the 5-week period. Nevertheless, DAI scores were notably raised in the chronic colitis group relative to the control group (P < 0.0001) after 5 weeks. Both acute and chronic colitis groups demonstrated no remarkable differences in terms of DAI scores (P = 0.9198). As shown in Fig. 1C, both forms of colitis were accompanied by significant shortening of the colon compared to their respective controls (P < 0.0001 for both), and the colon lengths did not vary significantly between the acute and chronic colitis models (P = 0.9724). Histopathological examination of the colon tissues of untreated control mice indicated clearly demarcated mucosa, intact epithelium, neatly arranged glands, abundant goblet cells, and absence of inflammatory cell infiltrates in the lamnia propria. In contrast, the DSS-treated mice exhibited considerable structural damage including incomplete glands, loss of epithelial cells, as well as extensive inflammatory cell infiltrates. Interestingly, the acute colitis group demonstrated notably thinned intestinal mucosa in contrast to that seen in the chronic colitis group. Mice with chronic colitis demonstrated locally thickened mucosa, which could be the result of alternating inflammatory and remission phases during the modeling process (Fig. 1E). Nevertheless, the histopathological scores of both groups were similar (P = 0.5113), and markedly higher compared to that of the respective controls (P < 0.0001 for both) (Fig. 1D).

DSS significantly induced inflammation in the colons of mice. A, B Disease activity index (DAI) dynamics across each cohort. C Mouse colon lengths of each group. D Colon histopathologic score of each group of mice. E Colonic tissues stained with H&E (200x; 400x). Data is depicted in terms of individual means or the mean ± SD of each group derived from three experimental replicates. ****P < 0.0001
Acute and chronic DSS exposure distinctly altered the diversity of the intestinal microflora
The intestinal microbiota profiles were analyzed by 16S rDNA sequencing. The Pielou index (Fig. 2A), which is indicative of species evenness, was notably lower in both the acute (P = 0.0022) and chronic (P = 0.0649) colitis groups compared to the respective controls, and did not differ significantly between the forms of colitis (P = 0.0022). The Shannon index (Fig. 2B) is a measure of species richness and evenness, and was decreased in both acute colitis (P = 0.0022) and chronic colitis (P = 0.0931) groups in contrast to the respective controls. Furthermore, acute colitis resulted in significantly lower Shannon index compared to chronic colitis (P = 0.0043). Taken together, DSS-induced colitis lowers mice gut microbiota species diversity, with more drastic changes seen in models of acute colitis.

DSS alters diversity of gut microbiota in mice models of colitis. A Pielou index. B Shannon index. α-diversity was derived using a combination of the species richness (species situation) and species evenness (distribution) using the Pielou and Shannon indices. C Gut microbiota relationships and distribution are characterized with a UPGMA clustering tree. Samples are represented by end branches. In general, samples belonging to similar groups were clustered into a large branch with different branches representing different groups. Distance is indicated by the vertical axis. Samples with similar structure of bacterial communities are located in the same branch. D NMDS (Non-metric Multi-Dimensional Scaling). NMDS representas a dimension reduction analysis established on distance ranking. It is superior to linear models (including PCA and PCoA) and more accurately translates the nonlinear structure of ecological data. The accuracy of the model is evaluated by the stress value. The smaller the stress value is, the more reliable the model is. Generally, the stress value less than 0.1 is better. **P < 0.01
The β-diversities of the intestinal microbiota across both cohorts were assessed using UPGMA and FastTree (Fig. 2C), NMDS (Fig. 2D), PCA (Fig. S1A), and PCoA (Fig. S1B). The results of UPGMA and FastTree showed that the acute colitis and control groups were further apart compared to the chronic colitis group and its corresponding control, indicating that acute colitis induced more significant changes in the intestinal microflora. The distance between acute and chronic colitis mice models was halfway between each colitis cohort and its respective control. Consistent results were obtained with NMDS, PCA and PCoA (stress< 0.1, PCA1 + PCA2 > 50%, PCoA1 + PCoA2 > 50%). The acute colitis group was the farthest from the rest, indicating that acute exposure to DSS induced significant changes in the gut microbial structure.
Acute and chronic DSS-triggered colitis resulted in a distinct microbiota profile
As shown in Fig. 3A, the relative abundance of Bacteroidetes (50.28% vs. 37.05%, P = 0.037), Proteobacteria (22.09% vs. 3.73%, P = 0.006), Epsilonbacteraeota (8.79% vs. 1.74%, P = 0.037) and Deferribacteres (0.31% vs. 0.15%, P = 0.337) were notably elevated in mice with acute colitis in comparison to their controls, while that of Firmicutes (17.44% vs. 52.23%, P = 0.004), Verrucomicrobia (0.03% vs. 0.22%, P = 0.004), Actinobacteria (0.45% vs. 1.66%, P = 0.055), Patescibacteria (0.4% vs. 0.44%, P = 0.631), Acidobacteria (0.0005% vs. 0.82%, P = 0.003) and Planctomycetes (0% vs. 0.64%, P = 0.002) were decreased. Likewise, chronic colitis was linked to a significant increase in the relative proportions of Bacteroidetes (57.95% vs. 36.77%, P = 0.004), Verrucomicrobia (18.99% vs. 3.07%, P = 0.010), Epsilonbacteraeota (3.12% vs. 0.93%, P = 0.006), Deferribacteres (0.27% vs. 0.15%, P = 0.078), Acidobacteria (0.02% vs. 0.01%, P = 0.935) and Planctomycetes (0.08% vs. 0.01%, P = 0.150), and a decrease in that of Firmicutes (15.12% vs. 51.51%, P = 0.004), Proteobacteria (3.50% vs. 4.76%, P = 0.423), Actinobacteria (0.67% vs. 2.11%, P = 0.025) and Patescibacteria (0.17% vs. 0.35%, P = 0.150) compared to the corresponding control.

DSS alters dominant gut microbiota structures across different levels in mice with colitis. Stacked bar plot depicts the phylum (A) / family (B) / genus (C) / species (D) structure of gut microbiota in each group of mice
At the family level (Fig. 3B), Bacteroidaceae (33.85% vs. 1.57%, P = 0.004), Enterobacteriaceae (18.03% vs. 0.09%, P = 0.004), Helicobacteraceae (8.79% vs. 1.74%, P = 0.037) and Desulfovibrionaceae (3.94% vs. 2.81%, P = 0.749) showed increased abundance in the acute colitis group compared to the control, and Lachnospiraceae (9.04% vs. 29.22%, P = 0.004), Muribaculaceae (6.39% vs. 18.74%, P = 0.004), Ruminococcaceae (5.44% vs. 20.33%, P = 0.004), Rikenellaceae (0.69% vs. 8.69%, P = 0.004), Akkermansiaceae (0.03% vs. 0.10%, P = 0.037) and Prevotellaceae (0.38% vs. 6.77%, P = 0.004) had lower abundance. Furthermore, chronic colitis was associated with a higher abundance of Muribaculaceae (19.99% vs. 16.15%, P = 0.078), Bacteroidaceae (14.01% vs. 0.65%, P = 0.004), Rikenellaceae (12.46% vs. 12.22%, P = 0.262), Akkermansiaceae (18.78% vs. 3.07%, P = 0.010), Prevotellaceae (7.49% vs. 3.02%, P = 0.025) and Helicobacteraceae (3.12% vs. 0.93%, P = 0.006), and lower abundance of Lachnospiraceae (5.88% vs. 35.21%, P = 0.004), Ruminococcaceae (5.28% vs. 11.21%, P = 0.004), Enterobacteriaceae (0.03% vs. 0.04%, P = 0.337) and Desulfovibrionaceae (2.76% vs. 4.46%, P = 0.200).
Microbiome genera which displayed notable elevations in the acute colitis group compared to the control (Fig. 3C) included Bacteroides (33.85% vs. 1.57%, P = 0.004), Escherichia-Shigella (17.30% vs. 0.06%, P = 0.004), Helicobacter (8.79% vs. 1.74%, P = 0.037) and Parabacteroides (8.09% vs. 0.02%, P = 0.004), while the abundance of Lachnospiraceae_NK4A136_group (1.6% vs. 10.79%, P = 0.004), Akkermansia (0.03% vs. 0.1%, P = 0.037), Alistipes (0.42% vs. 7.1%, P = 0.004), Alloprevotella (0% vs. 4.8%, P = 0.002), Rikenellaceae_RC9_gut_group (0.15% vs. 0.77%, P = 0.016) and Odoribacter (0.24% vs. 0.94%, P = 0.016) was lower in the acute colitis cohort. The relative abundance of Bacteroides (14.01% vs. 0.65%, P = 0.004), Akkermansia (18.78% vs. 3.07%, P = 0.010), Escherichia-Shigella (0.02% vs. 0.005%, P = 0.871), Helicobacter (3.12% vs. 0.93%, P = 0.006), Alloprevotella (4.39% vs. 2.52%, P = 0.262), Rikenellaceae_RC9_gut_group (9.06% vs. 1.52%, P = 0.004) and Parabacteroides (0.42% vs. 0.11%, P = 0.016) was higher in the chronic colitis group, while that of Lachnospiraceae_NK4A136_group (2.37% vs. 23.80%, P = 0.004), Alistipes (3.18% vs. 9.86%, P = 0.004) and Odoribacter (3.31% vs. 4.53%, P = 0.262) was lower.
At the species level (Fig. 3D), acute colitis increased the relative abundance of Bacteroides_acidifaciens (31.41% vs. 0.94%, P = 0.004), Parabacteroides_goldsteinii (7.91% vs. 0.005%, P = 0.004), Lactobacillus_murinus (0.14% vs. 0.12%, P = 1.000), Bacteroides_massiliensis_dnLKV3 (0.72% vs. 0.44%, P = 0.337), Bacteroides_caecimuris (1.09% vs. 0.04%, P = 0.004), Desulfovibrio_fairfieldensis (0.68% vs. 0.28%, P = 0.200) and Lachnospiraceae_bacterium_28–4 (0.33% vs. 0.25%, P = 0.423), and decreased that of Lactobacillus_gasseri (0.03% vs. 0.12%, P = 0.022), Alistipes_inops (0.14% vs. 0.24%, P = 0.150) and Lactobacillus_reuteri (0.08% vs. 0.16%, P = 0.150). The relative abundance of Bacteroides_acidifaciens (9.44% vs. 0.21%, P = 0.004), Parabacteroides_goldsteinii (0.03% vs. 0.002%, P = 0.031), Bacteroides_massiliensis_dnLKV3 (2.78% vs. 0.34%, P = 0.004), Bacteroides_caecimuris (1.23% vs. 0.08%, P = 0.004), Desulfovibrio_fairfieldensis (0.41% vs. 0.38%, P = 1.000), Alistipes_inops (0.36% vs. 0.22%, P = 0.150) and Lachnospiraceae_bacterium_28–4 (0.05% vs. 0.008%, P = 0.004) was higher in the chronic colitis group, while that of Lactobacillus_murinus (0.47% vs. 2.2%, P = 0.522), Lactobacillus_gasseri (0.14% vs. 0.68%, P = 0.037) and Lactobacillus_reuteri (0.34% vs. 0.36%, P = 1.000) was lower compared to the control group.
Acute and chronic DSS-induced colitis led to distinct changes in the intestinal microbiota
Welch’s t test was used to analyze the microbial changes induced by acute colitis and chronic colitis. There was a lower abundance of the phyla Firmicutes, Verrucomicrobia, Actinobacteria, Acidobacteria and Planctomycetes in the acute colitis group compared to the control (Fig. 4A and B). On the other hand, the chronic colitis group, demonstrated increased abundance of Bacteroidetes, Verrucomicrobia and Epsilonbacteraeota, and decreased proportions of Firmicutes and Actinobacteria. As shown in Fig. 4C and D, the relative proportion of the Bacteroidaceae family increased remarkably in the acute colitis group, and that of Lachnospiraceae, Muribaculaceae, Ruminococcaceae, Rikenellaceae, Prevotellaceae, Marinifilaceae and Christensenellaceae decreased compared to the control. Furthermore, Muribaculaceae, Bacteroidaceae, Akkermansiaceae, Prevotellaceae, Helicobacteraceae, Tannerellaceae and Peptostreptococcaceae were highly abundant in the chronic colitis group relative to the control, while Lachnospiraceae, Ruminococcaceae, Eggerthellaceae and Peptococcaceae showed low abundance.

DSS alters gut microbiota structure in acute and chronic colitis mice differently at the phylum and family levels. A DSS alters the microbial structure of acute colitis mice at the phylum level. B DSS alters the microbial structure of chronic colitis mice at the phylum level. C DSS alters the microbial structure of acute colitis mice at the family level. D DSS alters the microbial structure of chronic colitis mice at the family level
As shown in Fig. 5A and B, the relative abundance of the Bacteroides and Turicibacter families was increased, while that of Lachnospiraceae_NK4A136_group, Alistipes, Alloprevotella, Rikenellaceae_RC9_gut_group, Odoribacter, Ruminococcaceae_UCG-014, Ruminiclostridium_9, Oscillibacter, Intestinimonas, Ruminiclostridium, Rikenella, Ruminiclostridium_5, Butyricicoccus, Anaerotruncus, Ruminococcus_1, Muribaculum, Ruminococcaceae_UCG-005, Christensenellaceae_R-7_group, Ruminococcaceae_NK4A214_group, Ruminococcaceae_UCG-009, Ruminococcaceae_UCG-013, Marmoricola, Sphingomonas and Brevibacillus decreased significantly in the acute colitis group. In the chronic colitis group, the most abundant genera relative to the control group were Bacteroides, Akkermansia, Helicobacter, Parabacteroides, Erysipelatoclostridium, Turicibacter and Romboutsia, whereas Lachnospiraceae_NK4A136_group, Alistipes, Enterorhabdus, Prevotellaceae_UCG-001, Butyricicoccus, Ruminiclostridium_6, Muribaculum, Ruminococcaceae_NK4A214_group, Family_XIII_UCG-001 and Flavonifractor had low abundance. At the species level (Fig. 5C and D), acute colitis drastically elevated the relative proportion of Bacteroides_acidifaciens, and lowered that of Ruminococcus_flavefaciens, Lachnospiraceae_bacterium_COE1 and Clostridium_leptum. Chronic colitis increased the abundance of Bacteroides_acidifaciens, Bacteroides_massiliensis_dnLKV3, Bacteroides_caecimuris, Akkermansia_muciniphila and Oscillibacter_sp_1–3. Thus, the proportion of SCFAs-producing bacteria was elevated in the intestines of younger mice exposed to DSS over a longer duration. In contrast, acute colitis increased the abundance of conditional pathogens.

DSS alters gut microbiota structure in acute and chronic colitis mice differently at the genus and species levels. A DSS alters the microbial structure of acute colitis mice at the genus level. B DSS alters the microbial structure of chronic colitis mice at the genus level. C DSS alters the microbial structure of acute colitis mice at the species level. D DSS alters the microbial structure of chronic colitis mice at the species level
Discussion
DSS and 2,4,6-trinitro-benzenesulfonic acid (TNBS) are established agents for inducing acute and chronic colitis [17,18,19,20]. In this preliminary study, we found that administration of 3 and 2.5% DSS via drinking water can respectively induce acute and chronic colitis in mice while ensuring their survival. The affected mice showed significantly increased DAI scores, shortened colons, and significant damage to the intestinal mucosa. Interestingly, the weight of the control mice also fluctuated during the 5-week long cycle of chronic colitis modeling, which resulted in a transient increase in DAI score. However, the control group of the acute model was not affected due to the short treatment duration. Likewise, the colonic tissue of older mice also displayed limited neutrophil infiltration that did not penetrate beyond the lamina propria. In addition, the alternating active and remission phases during the induction of chronic colitis led to localized thickening of colonic mucosa, in contrast to the uniform thinning observed in acute colitis. Our models of acute and chronic colitis demonstrated good stability and homogeneity with a lack of significant variability in terms of disease features.
However, DSS-induced acute colitis led to notable reduction in the α-diversity of the gut microbiota compared to chronic colitis. Further distinctions between the two groups were highlighted with β-diversity analysis. Table S1 summarizes the changes in microbial structure at the levels of the phylum, family, genus and species. DSS-induced colitis stimulated the growth of Bacteroidetes, Epsilonbacteraeota and Deferribacteres, while lowering that of Firmicutes, Actinobacteria and Patescibacteria. The Firmicutes/Bacteroidetes ratio in the gut is an indicator of intestinal inflammation, and increases during colitis and other inflammatory conditions [21, 22]. In addition, the increased abundance of some conditional pathogens is also an important intestinal flora feature of UC. Li et al [23]. collected 56 mucosal microbiome samples from 28 Chinese UC patients and their healthy family partners, and identified four opportunistic “pathogens” (i.e., Clostridium tertium, Odoribacter splanchnicus, Ruminococcus gnavus and Flavonifractor plautii) with potential diagnostic and therapeutic significance for UC, which were inhibited in healthy individuals. Moreover, Huh et al. [24] identified Fusobacterium nucleatum as a marker for early gut microbial dysbiosis in inflammatory bowel diseases.
In our study, DSS-induced acute and chronic colitis simulated UC-associated microbial changes. Acute colitis raised the proportion of the Proteobacteria phylum and lowered that of the Verrucomicrobia phylum, whereas chronic colitis had the opposite effect. Proteobacteria is enriched in UC patients, which correlates with the significant reduction in intestinal SCFAs content (especially in the active phase) [25, 26]. In addition, the Verrucomicrobia phylum includes important SCFAs-producing bacteria such as Ruminococcus and Akkermansia. The abundance of the Bacteroidaceae and Helicobacteraceae families had increased, while that of Lachnospiraceae and Ruminococcaceae decreased in mice with acute and chronic colitis. The Helicobacteraceae family consists of conditional pathogens [27, 28], while Lachnospiraceae and Ruminococcaceae include SCFAs-producing bacteria that are negatively correlated with colitis [29,30,31]. Enterobacteriaceae and Desulfovibrionaceae were abundant in mice with acute colitis, while Rikenellaceae, Akkermansiaceae and Prevotellaceae showed lower abundance. Excessive proliferation of Enterobacteriaceae has been observed in the gut of UC patients [32, 33]. Surprisingly, the SCFAs-producing Rikenellaceae, Akkermansiaceae and Prevotellaceae were raised in mice with chronic colitis, which is contradictory to existing reports. Furthermore, both acute and chronic colitis were associated with a higher abundance of opportunistic pathogens (Bacteroides, Escherichia-Shigella, Helicobacter, Bacteroides_acidifaciens, Bacteroides_massiliensis_dnLKV3 and Bacteroides_caecimuris), and lower abundance of probiotics/SCFAs-producing bacteria (Lachnospiraceae_NK4A136_group, Alistipes, Butyricicoccus, Ruminococcaceae_NK4A214_group, Lactobacillus_gasseri and Lactobacillus_reuteri). Interestingly, the inhibition of SCFAs-producing bacteria was significantly stronger in mice with acute colitis compared to the chronic colitis model, especially for Rikenellaceae, Ruminococcaceae, Ruminiclostridium, Alistipes and Clostridium_leptum. These differences can be attributed to the longer modeling duration of chronic colitis as well as the higher concentration of DSS used to simulate acute colitis. High levels of DSS decreased food intake and resulted in more severe diarrhea and bloody stool symptoms, which lowered the impact of diet and fecal preference on the gut microbiota. Furthermore, the local thickening of the colonic mucosa during chronic colitis likely increased the proliferation of Akkermansia_Muciniphila, a myxotrophin bacterium related to mucous production and intestinal metabolism [34]. This organism is linked to a number of chronic conditions including diabetes and non-alcoholic steatohepatitis, and may be responsible for the aberrant mucin secretion during chronic colitis. Finally, Rikenellaceae, Ruminococcaceae, Ruminiclostridium, Alistipes and Clostridium_leptum thrive in an anaerobic environment, which is possibly disrupted during the induction of acute colitis, resulting in lower proliferation of these bacteria.
However, several limitations of the present study should be noted. First, the analysis of the acute colitis models can be performed within 1–2 weeks, whereas that of the chronic models may take 2–4 months [9]. In the present study, in order to ensure the survival rate of mice and effects of prolonged feeding on gut microbiota, we modeled colitis over a shorter period. Second, we did not assess the impact of different DSS concentrations and administration methods (such as oral gavage) on the intestinal microbiota. Third, we did not compare the effects of acute/chronic DSS exposure in different mouse strains (such as C57BL/6). Finally, the microbiota research method used in this study is relatively simple, and network analysis [23] of differential OTUs may adapt to reveal the complex relationships among microbes. Unfortunately, due to the small sample size of this study and our limited technical expertise in bioinformatics, we did not perform network analysis. However, these issues will be addressed in our subsequent study.
To summarize, both acute and chronic colitis stimulated changes in the gut microbiota, and the alterations in the acute colitis model are more consistent with that observed in UC patients. Furthermore, acute colitis inhibited the SCFAs-producing bacteria to a significantly greater extent compared to chronic colitis. Finally, younger age and shorter exposure to DSS are more conducive to the stability of the colitis microbiota in the mouse model. Our study provides an experimental basis for the analysis of colitis microbiota, and clearly shows that the acute colitis model simulates UC to a greater extent compared to the chronic colitis model.
Conclusions
DSS-induced acute and chronic colitis demonstrated similar symptoms and histopathological changes. However, the alterations in the acute colitis model are more consistent with that observed in UC patients than chronic colitis models. Acute colitis induced higher abundance of SCFAs-producing bacteria and lower α-diversity compared to chronic colitis.
Materials and methods
Establishment of DSS-induced colitis and treatment
This study was carried out in compliance with the ARRIVE guidelines. All animal experiment protocols were approved by the Animal Ethics Committee of Guangzhou First People’s Hospital (Approval number: 2017–202). Male BALB/c mice (n = 24, aged 6–8 weeks) were purchased from Guangdong Medical Laboratory Animal Center (Foshan, China, Certificate number SYXK 2013–0002). Animals were reared in a specific pathogen-free (SPF) environment at 24 °C under a 12-h light/dark cycle and relative humidity 50–70%. Mice were allowed free access to food and water. The mice were randomly divided into the acute blank (A_BLK), acute DSS-induced colitis (A_DSS), chronic blank (C_BLK) and chronic DSS-induced colitis (C_DSS) groups (n = 6 each). The control groups received regular drinking water for 1 week and 5 weeks respectively. Mice were stimulated to develop acute colitis through the addition of 3% DSS (MP Biomedicals, USA) into their drinking water for a week. Chronic colitis was induced by supplementing their drinking water with 2.5% DSS every other week for 5 weeks [8, 35]. Daily assessments of mice body weights, stool consistency, as well as the presence of blood-stained stool and anus were carried out. Mice were anesthetized with pentobarbital sodium and euthanized via transcardiac perfusion at the end of the experiments, and their colons were harvested, measured, and fixed overnight in 10% neutral buffered formalin. Stool samples were also collected from the cecum.
DAI scoring
A previously published protocol was adapted to assess the Disease Activity Index (DAI) [36] based on the following parameters: 1) weight loss, in terms of percentage from original body weight (0 – no loss; 1–1–5%; 2–5–10%; 3–10–15%; and 4 - > 15%), 2) stool consistency (0 - normal; 1 - pasty and not adherent to the anus; 2 - pasty and mildly adherent to the anus; 3 - pasty and adherent to the anus; and 4 - watery), and 3) rectal bleeding (0 - hemoccult (−); 1 - hemoccult (±); 2 - hemoccult (+); 3 - hemoccult (++); and 4 - obvious blood in stool) [37].
Histopathological evaluation
The entire length of the colon was dissected along the mesenteric border from each mouse, sliced longitudinally, and rinsed with cold saline. The tissue samples were fixed in buffered formalin, embedded in paraffin, and cut into 5 μm-thick sections. The slides were then stained with hematoxylin and eosin (HE) and scored independently by two investigators who were blinded to the grouping as follows: 1) inflammatory changes (0 - no inflammatory cells infiltrate; 1 – inflammatory infiltrates until the lamina propria; 2 – inflammatory infiltrates until the submucosa; and 3 - transmural infiltration), 2) presence of ulceration (0 - no ulceration; 1–1 ~ 2 ulcers; 2–3 ~ 4 ulcers; 3 - > 4 ulcers), 3) mucosal hyperplasia (0 - normal; 1 – mildly fibrosed and thickened mucosa; 2 – fibrous hyperplasia and mucosal thickening; 3 – extensive fibrous hyperplasia or granulation and mucosal thickening), and 4) edema (0 - none; 1–0 ~ 30%; 2–30 ~ 70%; 3 - > 70%) [38]..
Extraction of fecal DNA and analysis of gut microbiota
The HiPure Stool DNA Kit (Magen, Guangzhou, China) was used to extract total fecal DNA based on instructions provided by the manufacturer. The following primers were used to amplify the 16S rDNA target regions: 341F: CCTACGGGNGGCWGCAG; 806R: GGACTACHVGGGTATCTAAT [39]. Amplified products of 400–450 bpm were purified using the Phusion High-Fidelity PCR Master Mix (New England Biolabs, Beverly, MA, USA). Sequencing libraries were generated using the TruSeq DNA PCR-Free Sample Preparation Kit (Illumina, San Diego, CA, USA) based on protocols stipulated by the manufacturer before adding in index codes. The quality of the constructed library was evaluated using the Agilent Bioanalyzer 2100 system and Qubit@ 2.0 Fluorometer (Thermo Scientific, Carlsbad, CA, USA), an the Illumina HiSeq 2500 platform (Tianjin Novogene Bioinformatics Technology Co., Ltd) was used for sequencing [39].
Bioinformatics analyses
Quality control and reads assembly
Reads filtering
Raw data containing adapters or low-quality reads were further filtered according to the FASTP (version 0.18.0) criteria: 1) containing more than 10% of unknown nucleotides (N) and 2) containing less than 50% of bases with quality (Q-value)>20.
Reads assembly
Paired end clean reads were merged as raw tags using FLSAH (version 1.2.11) with a minimum overlap of 10 bp and mismatch error rates of 2%.
Raw tag filtering
Noisy sequences of raw tags were filtered by QIIME (version 1.9.1) pipeline under specific filtering conditions [40] to obtain the high-quality clean tags. The filtering conditions are as follows: 1) Break raw tags from the first low quality base site where the number of bases in the continuous low-quality value (the default quality threshold is <=3) reaches the set length (the default length is 3), and 2) filter tags with high-quality base length is less than 75% of the tag length.
In total, we obtained 1,344,698 tags without primers, with 56,029 tags per sample on average. [Link for the NCBI dataset: http://www.ncbi.nlm.nih.gov/bioproject/738354].
α-Diversity analysis
QIIME (version 1.9.1) was used to derive the α-diversity indices [41]. The ggplot2 package of the R project (version 2.2.1) was used to perform OTU rarefaction and plot rank abundance curves. Various α-indices (Pielou index and Shannon index) between two groups were calculated using the Wilcoxon rank test in the R project Vegan package (version 2.5.3). The Tukey’s HSD test was used to evaluate α-indices (Pielou index and Shannon index) among three or more groups.
Operational Taxonomic Units (OTUs) analysis and β-diversity analysis
Effective tags of OTUs ≥ 97% were clustered together with using UPARSE software (version 9.2.64) [42]. Each cluster was represented by the most abundant tag sequence of each cluster. The VennDiagram package [43] of the R project (version 1.6.16) was used to analyze element distribution, while the Vegan package in the R project (version 2.5.3) was utilized for principal component analysis (PCA). Relevant sequences were aligned with the software Muscle [44] (version 3.8.31) and clustering trees were constructed using the FastTree [45] (version 2.1) and UPGMA (Unweighted Pair Group Method with Arithmetic Mean) software. Multivariate statistical techniques including PCoA (principal coordinates analysis) [46] and NMDS (non-metric multi-dimensional scaling) [47] of Bray-Curtis distances were calculated using the Vegan package (version 2.5.3) and plotted using ggplot2 package (version 2.2.1).
Community composition analysis
A naive Bayesian model using RDP classifier [48] (version 2.2) based on SILVA [49] (version 132) or Greengene [50] (version gg_13_5) databases was used to classify sequences based on organisms. The community composition was visualized as a stacked bar plot using ggplot2 package [http://CRAN.R-project.org/package=ggplot2] (version 2.2.1) in the R project.
Welch’s t test
Welch’s t test was used to evaluate significant variances in terms of mean abundance of species between two groups. When there are two groups in the comparison group and total repeat samples in each group ≥3, the number of species tags/total tags of at least one sample ≥ 0.1%, and the top 1000 high abundance OTUs are statistically tested with R language vegan data package.
Statistical analysis
Data is depicted in terms of percentage or the mean ± standard deviation (SD). Inter- and intra-group differences were analyzed by the Wilcoxon signed rank test or unpaired t-test, one-way ANOVA and post hoc Tukey’s test as appropriate. The SPSS software (version 23.0; IBM Corp.) was utilized for all statistical analyses. Statistical significance was granted when the p values were < 0.05.
Availability of data and materials
The original contributions presented in the study are publicly available. These data can be found here: http://www.ncbi.nlm.nih.gov/bioproject/738354, BioProject ID is PRJNA 738354. Other datasets used and analyzed during the current study are available from the corresponding authors on reasonable request.
Abbreviations
- DSS:
-
Dextran sulfate sodium
- UC:
-
Ulcerative colitis
- DAI:
-
Disease activity index
- SCFAs:
-
Short chain fatty acids
- IBD:
-
Inflammatory bowel disease
- CD:
-
Crohn’s disease
- OTUs:
-
Operational taxonomic unit
- UPGMA:
-
Unweighted pair group method with arithmetic mean
- NMDS:
-
Non-metric multi-dimensional scaling
- PCA:
-
Principal component analysis
- PCoA:
-
Principal coordinates analysis
References
Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98(3):694–702. https://doi.org/10.1016/0016-5085(90)90290-h.
Randhawa PK, Singh K, Singh N, Jaggi AS. A review on chemical-induced inflammatory bowel disease models in rodents. Korean J Physiol Pharmacol. 2014;18(4):279–88. https://doi.org/10.4196/kjpp.2014.18.4.279.
Kiesler P, Fuss IJ, Strober W. Experimental models of inflammatory bowel diseases. Cell Mol Gastroenterol Hepatol. 2015;1(2):154–70. https://doi.org/10.1016/j.jcmgh.2015.01.006.
Spalinger MR, Atrott K, Baebler K, Schwarzfischer M, Melhem H, Peres DR, et al. Administration of the Hyper-immune Bovine Colostrum Extract IMM-124E ameliorates experimental murine colitis. J Crohns Colitis. 2019;13(6):785–97. https://doi.org/10.1093/ecco-jcc/jjy213.
Hernandez-Chirlaque C, Aranda CJ, Ocon B, Capitan-Canadas F, Ortega-Gonzalez M, Carrero JJ, et al. Germ-free and antibiotic-treated mice are highly susceptible to epithelial injury in DSS colitis. J Crohns Colitis. 2016;10(11):1324–35. https://doi.org/10.1093/ecco-jcc/jjw096.
Hoebler C, Gaudier E, De Coppet P, Rival M, Cherbut C. MUC genes are differently expressed during onset and maintenance of inflammation in dextran sodium sulfate-treated mice. Dig Dis Sci. 2006;51(2):381–9. https://doi.org/10.1007/s10620-006-3142-y.
Eichele DD, Kharbanda KK. Dextran sodium sulfate colitis murine model: an indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J Gastroenterol. 2017;23(33):6016–29. https://doi.org/10.3748/wjg.v23.i33.6016.
Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2(3):541–6. https://doi.org/10.1038/nprot.2007.41.
Wirtz S, Popp V, Kindermann M, Gerlach K, Weigmann B, Fichtner-Feigl S, et al. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat Protoc. 2017;12(7):1295–309. https://doi.org/10.1038/nprot.2017.044.
Augenlicht LH, Mariadason JM, Wilson A, Arango D, Yang W, Heerdt BG, et al. Short chain fatty acids and colon cancer. J Nutr. 2002;132(12):3804S–8S. https://doi.org/10.1093/jn/132.12.3804S.
Segain JP. Raingeard de la Bletiere D, Bourreille a, Leray V, Gervois N, Rosales C, et al. butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn's disease. Gut. 2000;47(3):397–403. https://doi.org/10.1136/gut.47.3.397.
Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol. 2011;17(12):1519–28. https://doi.org/10.3748/wjg.v17.i12.1519.
Miller TL, Wolin MJ. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl Environ Microbiol. 1996;62(5):1589–92. https://doi.org/10.1128/aem.62.5.1589-1592.1996.
Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294(1):1–8. https://doi.org/10.1111/j.1574-6968.2009.01514.x.
Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19(1):29–41. https://doi.org/10.1111/1462-2920.13589.
Li Z, Hu G, Zhu L, Sun Z, Jiang Y, Gao MJ, et al. Study of growth, metabolism, and morphology of Akkermansia muciniphila with an in vitro advanced bionic intestinal reactor. BMC Microbiol. 2021;21(1):61. https://doi.org/10.1186/s12866-021-02111-7.
Xu HM, Huang HL, Xu J, He J, Zhao C, Peng Y, et al. Cross-Talk Between Butyric Acid and Gut Microbiota in Ulcerative Colitis Following Fecal Microbiota Transplantation. Front Microbiol. 2021;12:658292. https://doi.org/10.3389/fmicb.2021.658292.
Xu HM, Zhou YL, Xu J, Li YF, Zhao C, Huang HL, et al. Inhibition of PD-1 protects against TNBS-induced colitis via alteration of enteric microbiota. Biomed Res Int. 2021;2021:4192451. https://doi.org/10.1155/2021/4192451.
Xu J, Xu HM, Peng Y, Zhao C, Zhao HL, Huang W, et al. The effect of different combinations of antibiotic cocktails on mice and selection of animal models for further microbiota research. Appl Microbiol Biotechnol. 2021;105(4):1669–81. https://doi.org/10.1007/s00253-021-11131-2.
Zhou Y, Xu H, Xu J, Guo X, Zhao H, Chen Y, et al. F. prausnitzii and its supernatant increase SCFAs-producing bacteria to restore gut dysbiosis in TNBS-induced colitis. AMB Express. 2021;11(1):33. https://doi.org/10.1186/s13568-021-01197-6.
Grigor'eva IN. Gallstone Disease, Obesity and the Firmicutes/Bacteroidetes Ratio as a Possible Biomarker of Gut Dysbiosis. J Pers Med. 2020;11(1). https://doi.org/10.3390/jpm11010013.
Pittayanon R, Lau JT, Leontiadis GI, Tse F, Yuan Y, Surette M, et al. Differences in gut microbiota in patients with vs without inflammatory bowel diseases: a systematic review. Gastroenterology. 2020;158(4):930–46 e1. https://doi.org/10.1053/j.gastro.2019.11.294.
Li W, Sun Y, Dai L, Chen H, Yi B, Niu J, et al. Ecological and network analyses identify four microbial species with potential significance for the diagnosis/treatment of ulcerative colitis (UC). BMC Microbiol. 2021;21(1):138. https://doi.org/10.1186/s12866-021-02201-6.
Huh JW, Roh TY. Opportunistic detection of Fusobacterium nucleatum as a marker for the early gut microbial dysbiosis. BMC Microbiol. 2020;20(1):208. https://doi.org/10.1186/s12866-020-01887-4.
Ma HQ, Yu TT, Zhao XJ, Zhang Y, Zhang HJ. Fecal microbial dysbiosis in Chinese patients with inflammatory bowel disease. World J Gastroenterol. 2018;24(13):1464–77. https://doi.org/10.3748/wjg.v24.i13.1464.
Zakerska-Banaszak O, Tomczak H, Gabryel M, Baturo A, Wolko L, Michalak M, et al. Dysbiosis of gut microbiota in polish patients with ulcerative colitis: a pilot study. Sci Rep. 2021;11(1):2166. https://doi.org/10.1038/s41598-021-81628-3.
Alkadhi S, Kunde D, Cheluvappa R, Randall-Demllo S, Eri R. The murine appendiceal microbiome is altered in spontaneous colitis and its pathological progression. Gut Pathog. 2014;6:25. https://doi.org/10.1186/1757-4749-6-25.
Ooi JH, Li Y, Rogers CJ, Cantorna MT. Vitamin D regulates the gut microbiome and protects mice from dextran sodium sulfate-induced colitis. J Nutr. 2013;143(10):1679–86. https://doi.org/10.3945/jn.113.180794.
Parada Venegas D, De la Fuente MK, Landskron G, Gonzalez MJ, Quera R, Dijkstra G, et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019;10:277. https://doi.org/10.3389/fimmu.2019.00277.
Peng L, Gao X, Nie L, Xie J, Dai T, Shi C, et al. Astragalin attenuates dextran sulfate sodium (DSS)-induced acute experimental colitis by alleviating gut microbiota Dysbiosis and inhibiting NF-kappaB activation in mice. Front Immunol. 2020;11:2058. https://doi.org/10.3389/fimmu.2020.02058.
Sasaki K, Inoue J, Sasaki D, Hoshi N, Shirai T, Fukuda I, et al. Construction of a model culture system of human colonic microbiota to detect decreased Lachnospiraceae abundance and Butyrogenesis in the feces of ulcerative colitis patients. Biotechnol J. 2019;14(5):e1800555. https://doi.org/10.1002/biot.201800555.
Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2(3):204. https://doi.org/10.1016/j.chom.2007.08.002.
Skuja V, Derovs A, Pekarska K, Rudzite D, Lavrinovica E, Piekuse L, et al. Gut colonization with extended-spectrum beta-lactamase-producing Enterobacteriaceae may increase disease activity in biologic-naive outpatients with ulcerative colitis: an interim analysis. Eur J Gastroenterol Hepatol. 2018;30(1):92–100. https://doi.org/10.1097/MEG.0000000000000989.
Zhai Q, Feng S, Arjan N, Chen W. A next generation probiotic, Akkermansia muciniphila. Crit Rev Food Sci Nutr. 2019;59(19):3227–36. https://doi.org/10.1080/10408398.2018.1517725.
Jiang Z, Mu W, Yang Y, Sun M, Liu Y, Gao Z, et al. Cadmium exacerbates dextran sulfate sodium-induced chronic colitis and impairs intestinal barrier. Sci Total Environ. 2020;744:140844. https://doi.org/10.1016/j.scitotenv.2020.140844.
Murthy SN, Cooper HS, Shim H, Shah RS, Ibrahim SA, Sedergran DJ. Treatment of dextran sulfate sodium-induced murine colitis by intracolonic cyclosporin. Dig Dis Sci. 1993;38(9):1722–34. https://doi.org/10.1007/BF01303184.
Rachmilewitz D, Karmeli F, Takabayashi K, Hayashi T, Leider-Trejo L, Lee J, et al. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology. 2002;122(5):1428–41. https://doi.org/10.1053/gast.2002.32994.
Lin H, Wang Q, Liu L, Chen Z, Das R, Zhao Y, et al. Colonization of mice with amoxicillin-associated Klebsiella variicola drives inflammation via Th1 induction and Treg inhibition. Front Microbiol. 2020;11:1256. https://doi.org/10.3389/fmicb.2020.01256.
Guo M, Wu F, Hao G, Qi Q, Li R, Li N, et al. Bacillus subtilis improves immunity and disease resistance in rabbits. Front Immunol. 2017;8:354. https://doi.org/10.3389/fimmu.2017.00354.
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013;10(1):57–9. https://doi.org/10.1038/nmeth.2276.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6. https://doi.org/10.1038/nmeth.f.303.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–8. https://doi.org/10.1038/nmeth.2604.
Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35. https://doi.org/10.1186/1471-2105-12-35.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. https://doi.org/10.1093/nar/gkh340.
Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490. https://doi.org/10.1371/journal.pone.0009490.
Shao H, Zhang C, Xiao N, Tan Z. Gut microbiota characteristics in mice with antibiotic-associated diarrhea. BMC Microbiol. 2020;20(1):313. https://doi.org/10.1186/s12866-020-01999-x.
Rakshit S, Ananthasuresh GK. An amino acid map of inter-residue contact energies using metric multi-dimensional scaling. J Theor Biol. 2008;250(2):291–7. https://doi.org/10.1016/j.jtbi.2007.09.032.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7. https://doi.org/10.1128/AEM.00062-07.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35(21):7188–96. https://doi.org/10.1093/nar/gkm864.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–72. https://doi.org/10.1128/AEM.03006-05.
Acknowledgments
Fei Yao is the wife of the first author (Hao-Ming Xu). Here we would like to thank Fei Yao for her support and encouragement over the past 10 years, whether in providing professional advice in data analysis or assisting Hao-Ming Xu in completing various difficult tasks. She is a kind-hearted woman, a competent wife and an excellent data analyst.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (81700487, 81970507), Guangdong Medical Science and Technology Research Fund (A2016340), Guangdong Planned Project of Science and Technology (2016A020215206), Guangzhou Planned Project of Science and Technology (201904010132, 202002020012, 202002030288, 202002030293) and Innovative Clinical Technique of Guangzhou (2019GX05).
Author information
Authors and Affiliations
Contributions
H.M.X. designed the study and drafted the manuscript; H.L.H. and Y.D.L. performed animal experiments, statistical analysis and interpretation of the data; J.Q.Z., J.X., H.L.Z., and X.G. participated in animal experiments, recorded general status of experimental animals, collected samples and related tests; Y.L.Z., H.T.C., and W.S. interpreted the data and revised the manuscript; Y.Q.N. and Y.J.Z. designed and organized the study, interpreted the data and revised the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All methods were carried out in accordance with relevant guidelines and regulations. All animal experiment protocols were approved by the Animal Ethics Committee of Guangzhou First People’s Hospital (Approval number: 2017–202), and all experiments were performed in compliance with the ARRIVE guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
DSS alters β-diversity (PCA & PCoA) of gut microbiota in mice models of colitis. (A) PCA (Principal Component Analysis). The PC1 coordinate indicates the primary principal component while its contribution value to sample variability is depicted in percentage in brackets. The PC2 coordinate represents the second principal component while its contribution value to sample variability is depicted in percentage in brackets. Samples are represented by colored dots in the panel. Sample compositions which are similar are located more closely together on the PCA diagram. Samples of various different environments signify their respective aggregation distribution. (B) PCoA (Principal Co-ordinate Analysis). PCoA is a dimension reduction analysis based on the distance matrix, and the percentage (the number in brackets of the axis title) is used to evaluate the explanation degree of each coordinate axis to the overall difference of microbial structure. Generally, it is better that the sum of PCoA1 and PCoA2 is more than 50%. Table S1. Alterations of microbial structure at different levels in acute / chronic colitis mice.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, HM., Huang, HL., Liu, YD. et al. Selection strategy of dextran sulfate sodium-induced acute or chronic colitis mouse models based on gut microbial profile. BMC Microbiol 21, 279 (2021). https://doi.org/10.1186/s12866-021-02342-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-021-02342-8