
Abstract
Background
Clostridioides difficile infection (CDI) has a high recurrent infection rate. Faecal microbiota transplantation (FMT) has been used successfully to treat recurrent CDI, but much remains unknown about the human gut microbiota response to replacement therapies. In this study, antibiotic-mediated dysbiosis of gut microbiota and bacterial growth dynamics were investigated by two quantitative methods: real-time quantitative PCR (qPCR) and direct culture enumeration, in triple-stage chemostat models of the human colon. Three in vitro models were exposed to clindamycin to induce simulated CDI. All models were treated with vancomycin, and two received an FMT. Populations of total bacteria, Bacteroides spp., Lactobacillus spp., Enterococcus spp., Bifidobacterium spp., C. difficile, and Enterobacteriaceae were monitored using both methods. Total clostridia were monitored by selective culture. Using qPCR analysis, we additionally monitored populations of Prevotella spp., Clostridium coccoides group, and Clostridium leptum group.
Results
Both methods showed an exacerbation of disruption of the colonic microbiota following vancomycin (and earlier clindamycin) exposure, and a quicker recovery (within 4 days) of the bacterial populations in the models that received the FMT. C. difficile proliferation, consistent with CDI, was also observed by both qPCR and culture. Pearson correlation coefficient showed an association between results varying from 98% for Bacteroides spp., to 62% for Enterobacteriaceae.
Conclusions
Generally, a good correlation was observed between qPCR and bacterial culture. Overall, the molecular assays offer results in real-time, important for treatment efficacy, and allow the monitoring of additional microbiota groups. However, individual quantification of some genera (e.g. clostridia) might not be possible without selective culture.
Similar content being viewed by others


Background
Clostridioides difficile infection (CDI) is a healthcare-associated infection related with high morbidity, mortality and costs [1]. Vancomycin and fidaxomicin are the recommended antibiotics for the treatment of moderate to severe CDI [2], but recurrent infection rates can be as high as 20–30% [1, 3,4,5]. Faecal microbiota transplantation (FMT) has been used successfully as treatment for recurrent CDI (rCDI), with reported resolution rates that can reach ~ 90% [6,7,8]. However, much remains unknown about the human gut microbiota composition and behaviour, particularly in undefined microbiota replacement therapy, such as FMT.
The chemostat model has been validated against the intestinal contents of sudden-death victims and consists on a reliable representation on the microbial content and bacterial activities of the human colon [9]. This model of the human colon has been used to investigate the propensity of different antibiotics to induce CDI and the efficacy of treatments [10,11,12,13,14,15,16]. The results observed in the gut model have shown to correlate well with phase three clinical trials [10, 15], underlining the clinical relevance of this system. Bacteroides spp., Bifidobacterium spp. and Clostridium spp., are examples of bacterial genera highly abundant in the human gut that are often disrupted by antimicrobial therapy, as shown by ex vivo and in vitro studies [6, 13,14,15, 17,18,19,20,21]. The decline of microbial populations creates a potential niche for C. difficile spore germination, cell proliferation and toxin production, leading to CDI [10]. In the previous gut model studies, variations in microbial populations were investigated using bacterial culture. The accuracy of the gut model studies predicting clinical outcomes suggests that the bacterial populations targeted provide a suitable representation of the healthy human microbiota [18]. However, some important groups of the human gut microbiota are difficult to monitor by this method due to taxonomic complexity. Furthermore, bacterial culture is demanding on staff time and requires between 24 h and 48 h of plate incubation for colony growth, which can delay interventions to the model; e.g. commencement of antibiotic course during simulated CDI. Similar to bacterial culture, real-time PCR methodology can be used to investigate gut microbiota variations from a quantitate perspective [20,21,22,23,24], but populations targeted by this method may differ from those investigated by direct culture. Taxonomic profiling using 16S sequencing can also be used to increase our understanding of the gut microbiota populations exposed to antibiotic pressure [6, 19, 25]. However, the variations observed by 16S sequencing are qualitative, as bacterial groups differ in the number of 16S gene copies encoded on their genomes. Additionally, gut microbiota exposure to antibiotics can create ‘artefacts’, as the proportion of reads assigned to a bacterial population may increase as result of the depletion of abundant bacterial populations, and not due to bacterial expansion.
To better understand the dynamics of bacterial populations during replacement therapy and infection, we investigated the gut microbiota reconstitution following a simulated FMT, alongside an experiment of rCDI, using in vitro chemostat models of the human colon. Direct enumeration of key bacterial groups was monitored using two quantitative methodologies: selective bacteriological agars, and real-time quantitative PCR (qPCR). The advantages and disadvantages of each methodology, when applied to studies of the human gut microbiota, is investigated.
Results
Bacterial populations monitored by qPCR and bacterial culture
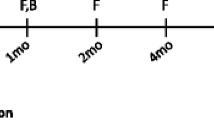
Gut microbiota variations were monitored in three chemostat models of the human colon, here described as A, B and C. Project outline is shown in Fig. 1. All models were started with the same human faecal slurry and underwent clindamycin induced CDI, followed by treatment with vancomycin. Up to the end of vancomycin treatment all models experienced the same experimental design, representing technical replicates. Bacterial populations were monitored in vessel 3 of model A, B and C throughout the experiment, using bacterial culture and qPCR assays. As the bacterial populations showed minor variations, only the mean logarithm values of the three models is shown. Following vancomycin instillation, model A was left without additional treatment, representing simulated rCDI. Three days post completion of vancomycin treatment, models B and C received FMT therapy. This three-day period allowed the washout of vancomycin to prevent residual antibiotic effects on the transplanted microbiota. The post-FMT results are shown as the mean of model B and C data, or model A only.

Experimental timeline for the recurrent CDI model A, and the faecal microbiota transplantation (FMT) models B and C. All models followed the same experimental design to induce and treat simulated CDI with clindamycin and vancomycin, respectively. Following treatment course with vancomycin, model A received no further intervention (red arrow), whereas model B and C received an FMT three days post vancomycin treatment (purple arrow). Black lines indicate the times at which samples were collected for qPCR and culture analysis
Real-time PCR assays species- and group-specific were applied to DNA extracted from gut model samples at key stages of the experiment to monitor population dynamics. Furthermore, total bacteria, Bacteroides spp., Lactobacillus spp., Enterococcus spp., Bifidobacterium spp., and C. difficile were also monitored using culture assay.
qPCR and bacterial culture analyses showed that the total bacterial populations remained stable throughout the experiment except for a decline of ~ 1 log10 copies/μL and ~ 1 log10 cfu/mL, respectively, observed following clindamycin dosing. The qPCR data showed this decline to be significant (p < 0.005) (Fig. 2a). qPCR data also showed a significant (p < 0.005) increase in total bacteria following gut microbiota reconstitution by FMT in models B and C. For the Bacteroides spp. population (Fig. 2b), clindamycin instillation caused a significant decline of ~ 1 log10 copies/μL (p < 0.0005), followed by recovery (p < 0.005) during CDI phase. Vancomycin dosing led to a ~ 4 log10 copies/μL decline (p < 0.005). In model A, Bacteroides spp. recovered only by the end of the experiment, whereas in model B and C an increase of ~ 3.5 log10 copies/μL (p < 0.0005) was observed four days after FMT. Bacterial culture results showed similar variations in Bacteroides spp., namely a ~ 2 log10 cfu/mL decline was observed following clindamycin dosing and ~ 7 log10 cfu/mL decline was observed following vancomycin instillation, with a faster recovery to steady state levels being observed in model B and C. Analysis by qPCR showed significant declines in Lactobacillus spp. (p < 0.005) (Fig. 2c), and Enterococcus spp. (p < 0.0005) (Fig. 2d) populations during CDI period, followed by a further decline caused by vancomycin instillation (p < 0.0005). Culture populations of Lactobacillus spp. and Enterococcus spp. remained stable up to vancomycin dosing that caused a ~ 1.0 log10 cfu/mL decline in both populations. In model B and C, both qPCR and culture data showed a recovery of Enterococcus spp. populations to steady state levels following FMT.

Mean gut microbiota populations of (a) total bacteria, (b) Bacteroides spp., (c) Lactobacillus spp., (d) Enterococcus spp., and (e) C. difficile in vessel 3 of model A, B and C at the different stages of the experiment. Bars represent the levels in log10 copies/μL measured by qPCR, and lines represent the populations levels in log10 cfu/mL measured by bacterial culture. CD, C. difficile; rCDI, recurrent CDI; FMT, faecal microbiota transplantation. Asterisks represent significant variations by qPCR between time points: *correspond to p < 0.05, **correspond to p < 0.005, and ***correspond to p < 0.0005
Bifidobacterium spp. populations analysed by qPCR were stable prior to clindamycin instillation, which caused a ~ 1 log10 copies/μL (p < 0.005) decline to ~ 7 log10 copies/μL, followed by recovery to steady state levels during CDI (p < 0.005). Vancomycin instillation also caused a decline of ~ 1.5 log10 copies/μL (p < 0.0005). No significant variations were observed in model B and C following FMT. In all models, Bifidobacterium spp. populations recovered by the end of the experiment. Culture-based analysis showed a depletion of Bifidobacterium spp. during clindamycin (~ 6 log10 cfu/mL) and vancomycin to below the culture assay limit of detection. In model A, Bifidobacterium spp. remained undetected by culture until the end of the experiment. Following FMT, Bifidobacterium spp. increased ~ 2.5 log10 cfu/mL in model B and C and recovered to steady state levels (~ 8 log10 cfu/mL) by the end of the experiment.
C. difficile was investigated using qPCR assays for the 16S gene and the housekeeping gene gluD (Fig. 2e and Additional file 1: Figure S1). In both assays, C. difficile copy number declined ~ 1.5 log10 copies/μL (p < 0.0005) during clindamycin instillation. This was followed by ~ 2.2 log10 copies/μL increase (p < 0.0005) post antibiotic period, consistent with C. difficile cell proliferation in the gut models, and was accompanied by toxin production, corresponding to simulated CDI. Average toxin value prior to vancomycin treatment was 2 relative units (RUs). Vancomycin treatment caused a decline in C. difficile populations of ~ 3 log10 copies/μL (p < 0.0005) and toxin declined to undetectable levels. In model A, a ~ 1.4 log10 copies/μL increase in C. difficile was observed towards the end of the experiment (p < 0.05). This was accompanied by toxin production corresponding to 3 RUs, which is consistent with rCDI. Following the FMT in models B and C, no significant increase in C. difficile copy number was observed and no toxin was detected, suggesting resolution of CDI (Fig. 2e). Monitoring of C. difficile by culture showed a ~ 2 log10 cfu/mL decline in C. difficile counts following clindamycin dosing. C. difficile population then increased ~ 3 log10 cfu/mL consistent with cell proliferation. C. difficile declined to below the limit of detection with vancomycin instillation and remained low up to the end of the experiment when a sudden increase consistent with spore germination and cell proliferation, was observed in model A only.
Gut microbiota populations monitored by qPCR
Prevotella spp. and C. leptum are important bacterial populations in the human microbiome [20], however; these groups could only be monitored by qPCR due to lack of selective agars available (Fig. 3). Prevotella spp. and C. leptum did not significantly change following the addition of C. difficile spores. Clindamycin caused a ~ 1 log10 copies/μL (p < 0.0005) decline in Prevotella spp., followed by recovery during CDI phase (p < 0.005) (Fig. 3a). Vancomycin treatment also caused a decline of ~ 4 log10 copies/μL (p < 0.0005). In model A, Prevotella spp. recovered by the end of the experiment. In models B and C, the FMT led to a ~ 4 log10 copies/μL (p < 0.0005) recovery within 4 days. Although a decline in C. leptum was observed during clindamycin instillation, this was not significant (Fig. 3b). Vancomycin dosing caused a decline of ~ 3 log10 copies/μL (p < 0.0005). Full recovery of this population was observed at the end of the experiment for all models, but in model B and C, population level increased earlier, showing a ~ 1 log10 copies/μL (p < 0.0005) recovery just 4 days post-FMT.

Gut microbiota populations of a) Prevotella spp., and (b) C. leptum in vessel 3 of model A, B and C, investigated by qPCR only. CD, C. difficile; rCDI, recurrent CDI; FMT, faecal microbiota transplantation. Asterisks represent significant variations between time points: *correspond to p < 0.05, **correspond to p < 0.005, and ***correspond to p < 0.0005
Individually monitoring of selected populations
In this study, the qPCR results for C. coccoides were correlated with the culture data for Clostridium spp.. Among the primer pairs used in this study, the C. coccoides primers showed the higher specificity for the Clostridium species most commonly isolated from the gut model, being able to amplify 9 out of the 13 species investigated (Table 1). In addition to Clostridium species [18, 25], C. coccoides group includes species from several other genus. Due to its complexity, the populations that constitute the C. coccoides group were monitored by qPCR (Fig. 4a). Declines in C. coccoides, of ~ 1 log10 copies/μL (p < 0.005) and ~ 2 log10 copies/μL (p < 0.0005), were observed as a result of clindamycin and vancomycin dosing, respectively. In model B and C, this population recovered ~ 1.5 log10 copies/μL (p < 0.0005) post-FMT. Selective culture allowed the monitoring of several commensal clostridia species of the human microbiota (Fig. 4b). In this study, selective culture showed declines in total clostridia counts following clindamycin (~ 2 log10 cfu/mL) and vancomycin (~ 5 log10 cfu/mL). In model A, clostridia populations recovered within 4 weeks, whereas in model B and C a ~ 2 log10 cfu/mL increase was observed 4 days post-FMT. Clindamycin instillation caused an increase in both Enterobacteriaceae populations monitored by qPCR (~ 1 log10 copies/μL, p < 0.0005) (Fig. 4c) and lactose-fermenting Enterobacteriaceae monitored by selective culture (~ 1 log10 cfu/mL) (Fig. 4d). qPCR analysis showed a decline of ~ 1 log10 copies/μL in Enterobacteriaceae following vancomycin instillation (p < 0.0005); however, data of direct culture showed that lactose-fermenting Enterobacteriaceae populations increased ~ 1 log10 cfu/mL during this stage. According to qPCR, the FMT did not significantly affect Enterobacteriaceae populations. However, culture data showed a decline of ~ 1 log10 cfu/mL in lactose-fermenting populations in model B and C, 4 days post-treatment.

Levels of bacterial populations in vessel 3 of model A, B and C. (a) qPCR results for C. coccoides group in log10 copies/μL, (b) culture results in log10 cfu/mL for total clostridia, (c) qPCR results for Enterobacteriaceae in log10 copies/μL, (d) culture results in log10 cfu/mL for lactose-fermenting Enterobacteriaceae. CD, C. difficile; rCDI, recurrent CDI; FMT, faecal microbiota transplantation. Asterisks represent significant variations by qPCR between time points: *correspond to p < 0.05, **correspond to p < 0.005, and ***correspond to p < 0.0005
Correlation between bacterial culture and qPCR assays
A Pearson correlation coefficient was used to determine the relationship between bacterial populations enumerated by qPCR measurements and culture-based methods (Table 2). The higher correlation between methods was observed for bacterial genus/species targeted by both methods. This was particularly seen in Bacteroides spp., as the bacterial levels detected throughout the study showed a correlation of 98% between methods, suggesting these assays target the same Bacteroides populations within the gut models. Good correlations were also observed for Lactobacillus spp., Enterococcus spp. and Bifidobacterium spp., corresponding to 94, 85 and 73%, respectively. The linear relationship between methods for all these populations was > 0.85. As the models are maintained in anaerobic conditions, for the correlation analysis, the qPCR data targeting total Eubacteria was compared the counts of total bacteria cultured in anaerobic conditions, with an association of 86% between assays. Unsurprisingly, given the slightly different targets of qPCR and culture enumeration, the lower correlation values were observed for Enterobacteriaceae (62%) and clostridia (63%), with both assays showing a linear relationship of 0.79. These correlation values were highly affected by the results observed in each assay during antibiotic instillation. The correlation analysis for Enterobacteriaceae showed the most variation during vancomycin dosing. Similarly, Clostridium spp. direct enumeration and C. coccoides qPCR assay showed the most variation during clindamycin dosing, which declined the linear correlation between assays from 0.91 to the reported 0.79.
Discussion
Gut microbiota variations and microbiota reconstitution were monitored in three chemostat in vitro models of the human colon using two methods of quantitative bacterial enumeration: culture-based assay and qPCR. Models A, B and C underwent clindamycin induced CDI followed by treatment with vancomycin. In model A, CDI was treated with vancomycin only, whereas in models B and C vancomycin treatment was followed by an FMT. Post-treatment, bacterial populations were allowed four weeks to recover.
Similar to previous in vitroEscherichia coli cells gut model studies [11,12,13,14] antibiotic therapy with clindamycin and vancomycin caused a decline in several of the bacterial populations monitored, particularly Bifidobacterium spp., Bacteroides spp., clostridia, Enterococcus spp., Lactobacillus spp., and C. difficile. Disruption of the gut microbiota was exacerbated following vancomycin dosing, with these declines being observed by both bacterial culture and qPCR.
A good correlation (> 85%) between qPCR and direct culture was observed for most populations investigated. The lowest correlation value, 62%, was observed between the qPCR assay for Enterobacteriaceae and selective culture for lactose-fermenting Enterobacteriaceae populations, partially due to the different trends observed between assays during vancomycin dosing. Escherichia spp., Klebsiella spp., Citrobacter spp., and Enterobacter spp. species are among the genera of the normal intestinal flora of humans that have the capacity to process lactose [27]. However, some genera within the Enterobacteriaceae family are non-lactose fermenters and may require other media for its selective culture [27, 28]. Among these, Salmonella spp., Shigella spp., Serratia spp., and Proteus spp. species can be part of the human gastrointestinal microbiota, and their presence was confirmed in these gut models by MALDI-TOF identification of selected colonies in non-selective nutrient agar. As the qPCR reaction targets the 16S region of the overall Enterobacteriaceae gut populations without distinguish the lactose fermenting species, this possibly explains the lower correlation observed in this case between qPCR and bacterial culture.
Prevotella spp., C. coccoides and C. leptum populations were investigated due to their importance for a healthy microbiome [17, 20, 21, 25]. We found that direct enumeration of Clostridium spp. correlated with the qPCR results for C. coccoides based on the specificity of this primer pair for clostridia species commonly isolated from gut model samples; however, not all Clostridium species were amplified. Correlation between culture of Clostridium spp. and the qPCR assay for C. coccoides mostly differed during clindamycin dosing period, when culture showed a high decline in clostridia counts that was less noticeable by qPCR. Within the Clostridium genus, C. coccoides (Clostridium cluster XIVa) and C. leptum (Clostridium cluster IV) are two highly predominant groups of the human gastrointestinal tract [17, 18, 20, 25]. Each of these groups is complex, with C. cocoides being closer in composition to the Lachnospiraceae family and C. leptum to the Ruminococcaceae family. C. coccoides includes Butyrivibrio, Clostridium, Coprococcus, Dorea, Eubacterium, Lachnospira, Roseburia, and Ruminococcus species, and C. leptum includes Clostridium, Ruminococcus, Eubacterium and Faecalibacterium prausnitzii species [18, 19, 25, 29]. Since several bacterial populations are represented in the C. coccoides group, it is likely that some species within the group were less disturbed by clindamycin instillation, which was reflected in the qPCR assay results.
In this study, C. difficile was investigated using two qPCR assays, one for the species 16S region and other for the housekeeping gene gluD. Results consistently showed C. difficile levels ~ 1 log10 copies/μL higher in the 16S gene assay compared to the gluD assay. Concentrations of C. difficile genomic DNA ranging from 12.5 ng to 0.024 ng were tested with both primer sets, showing results ~ 1.5 log10 copies/μL higher in the 16S assay compared to gluD. As C. difficile genome has 11 copies of the 16S rRNA operon and 1 copy of the gluD gene [30] this could possibly explain the apparent higher sensitivity of the 16S qPCR assay.
Overall, both quantitative analyses of gastrointestinal bacterial populations showed an earlier reconstitution of the gut microbiota in the models that received an FMT. Although both assays provide absolute quantification data, they have different limits of detection. As example, Bifidobacterium spp. was periodically undetectable by direct culture, whereas all commensal populations remained detected by qPCR throughout the experiment. Although the assays showed a good correlation, an improvement is likely to be observed if the limits of detection of both methodologies are taken into consideration. qPCR allowed us to monitor bacterial groups that would otherwise not be possible to investigate and offered a shorter turnaround time (less than 24 h) for results compared with culture. In addition to its high sensitivity, qPCR also allowed analysis of higher number of technical replicates adding the possibility of statistical analysis. However, by detecting DNA, we are unable to distinguish between viable and non-viable cells. Despite having a slower turnaround time, particularly for anaerobic populations that require 48 h incubation, the culture results reflect cell viability and coupled with MALDI-TOF identification, provided additional quantitative information on the most abundant bacterial species in the gut model at a given time. This allows for a better understanding of any specific pathways that may be active and their potential relevance in disease. Studies involving monitoring of multiple populations by culture assay can be challenging, however, for the phylogenetically inconsistent clostridia genus [29], the use of a single selective medium in this study allowed the monitoring of high abundant Clostridium species. Additionally, with bacterial culture, we were able to monitor C. difficile at different stages of the bacteria life cycle, by differentiate spores and vegetative cells which was not possible using qPCR.
Conclusions
Overall, the commensal bacterial populations monitored in this study showed a quicker recovery in the models that received an FMT, compared to treatment with vancomycin only. Alongside this recovery, the models that received an FMT did not show recurrent infection, whereas model A showed C. difficile proliferation and toxin production, consistent with rCDI. These results show the potential of replacement therapies in the reconstitution of the normal human microbiota. Furthermore, the qPCR assays correlated well with the results of bacterial enumeration by culture, with both methods showing antibiotic-mediated depletion of the gut microbiota populations, followed by reconstitution of gastrointestinal populations within four weeks. The molecular assays can potentially provide information on gut bacterial variations in real-time, reducing the standard requirements for bacterial culture, and allowing the monitoring of bacterial groups that would otherwise not be possible. However, in some cases it may not be possible to monitor an individual genus without selective culture.
Methods
Chemostat model assembly and experimental design
Three triple-stage chemostat models, assembled as previously described [10, 15] were run simultaneously. Briefly, each model consists of three glass vessels displayed in a weir cascade and kept at 37 °C by a water jacket. This system simulates the nutrient availability and pH conditions of the proximal, medial and distal colon in vessel 1, 2 and 3, respectively. The models are continuously fed with a complex nutritive medium (Table 3), at the pre-established dilution rate of 15 mL/h and an anaerobic environment was maintained by sparging the system with nitrogen. All models were inoculated with a slurry of pooled human faeces [10% w/v in pre-reduced phosphate buffered saline (PBS)] from 22 healthy volunteers, > 60 years of age, and with no history of antibiotic therapy in the previous 6 months. In each model, bacterial populations were allowed to equilibrate for 4 weeks, followed by addition of two doses of 107 cfu/mL C. difficile spores, a week apart. C. difficile spores of the PCR ribotype 027 strain 210 were prepared as described before [31], and added to the model. CDI was induced following clindamycin instillation (33.9 mg/L, four times daily for 7 days) (Fig. 1). C. difficile germination, growth and toxin production were monitored and at peak toxin production, all models were dosed with vancomycin (125 mg/L, four times daily for 7 days). Antibiotic treatments were based on previously reported faecal concentrations and prescribing recommendations for these drugs [2, 13]. Three days post vancomycin, model A was left without further intervention, whereas models B and C were treated with an FMT preparation. Up to the end of vancomycin treatment, all models followed the same experimental design, representing replicates for simulated CDI and treatment, as previously described [12,13,14]. Afterwards, model A proceeded as a representation of a simulated rCDI case, whereas model B and C proceeded as replicates of a simulated FMT treatment. The FMT experiment was performed in duplicate to confirm method reproducibility in the chemostat model. FMT was prepared similar to the initial slurry, by performing a 10-fold dilution with pre-reduced PBS of a faecal sample donated by a relative of one of the initial donors, aged 30, without history of antibiotic use in the previous 6 months. The FMT (50 mL) was added to the bottom of vessel 1 of models’ B and C at a 50 mL/h rate using a peristaltic pump. Microbial populations were monitored for 4 weeks post instillation in all models.
Bacterial culture and cytotoxin assay
Models were sampled for culture profiling of key intestinal microbiota populations using selective and non-selective agars described in Table 3. Populations of total bacteria, Clostridium spp. lactose-fermenting Enterobacteriaceae, Enterococcus spp., Bacteroides spp., Bifidobacterium spp., Lactobacillus spp., C. difficile total viable cells and C. difficile spores were monitored at the time points illustrated in Fig. 1. All bacterial populations and C. difficile spores were cultured as previously described [10]. Briefly, C. difficile spores were isolated by treating 0.5 mL of gut model fluid with 0.5 mL of 96% ethanol. The samples were incubated at room temperature for 1 h, serially diluted to 10− 3 in peptone water, and 20 μL of each sample dilution was plated in triplicate onto supplemented Braziers CCEY agar (Table 3). Plates were incubated anaerobically for 48 h and distinctive colonies were enumerated. Identification of bacterial isolates were confirmed by Matrix-Assisted Laser Desorption Ionization-Time of Flight (MALDI-TOF). Vessel 3 was particularly investigated due to its high microbial content [32] and because it represents the area of the colon more physiologically relevant for CDI [33]. The limit of detection for culture assay was ~ 1.22 log10 cfu/mL.
The C. difficile toxin levels were measured by cell cytotoxicity assay, as previously described [10]. Briefly, model fluid was centrifuged, filtered, and serially diluted on a 10-fold series. Each dilution was inoculated onto a confluent monolayer of Vero cells and incubated for 48 h at 37 °C at 5% CO2. Samples were considered positive for C. difficile toxin when > 70% cell rounding was observed. Results are expressed as RUs, as follows: positive result on a 1:10 dilution = 1 RU, 1:100 = 2 RU, etc.
DNA extraction of gut model samples
For molecular analysis, samples were taken from vessel 3 of each model as outlined in Fig. 1. For each time point, DNA extraction was performed in triplicate using the FastDNA Spin kit for Soil (Mpbio) following manufacturer’ instructions except that sample homogenisation was performed using Precellys 24 (Bertin Instruments) at 6500 rpm for 40 s. DNA concentration was measured using a Nanodrop 2000c and each sample was normalised to 5 ng/μL.
Preparation of control curves using plasmid standard DNA
For the analysis of Enterococcus spp., Bifidobacterium spp., Lactobacillus spp., and C. difficile, plasmids containing 16S gene inserts specific for these populations were prepared as described before [24]. Group-specific PCR reactions were performed on appropriate bacterial strains using the primers described in Table 4. The products were cloned using the TOPO TA cloning kit (Thermo Fisher Scientific) and inserts were verified by Sanger sequencing capillary electrophoresis (Applied Biosystems). Bacterial group specificity of the insert in transformed colonies was confirmed by blastn search and plasmid DNA was isolated using QIAprep spin miniprep kit (Qiagen). Plasmids containing 16S gene inserts for total bacteria, Enterobacteriaceae, Bacteroides spp., Prevotella spp., Clostridium coccoides and Clostridium leptum were provided by Dr. Cheleste M. Thorpe (Tufts Medical Center, USA) [24]. Plasmids were re-suspended, propagation was performed using TOP10 chemically competent Escherichia coli cells (Thermo Fisher Scientific) according to manufacturers’ instructions, and plasmid DNA was obtained as above. The molecular weight of each plasmid containing 16S gene inserts was determined using a web calculator (http://www.encorbio.com/protocols/Nuc-MW.htm). Purified plasmid concentrations were determined using Qubit 2.0 Fluorometer and 10-fold dilution series ranging from 5 × 109 copies/μL to 500 copies/μL were prepared.
qPCR assays and data analysis
The DNA levels of each bacterial genus/species were assessed using primers previously validated for qPCR (Table 4). Reactions containing final concentration of SYBR Green 1x Master Mix (Qiagen), 0.3 μM primers and 12.5 ng of DNA template were prepared to a final volume of 10 μL. A FAM-tagged probe at 0.25 μM was added to the Eubacteria reaction mix [24]. Reactions were analysed in a 7500 Real-Time PCR System (Thermo Fisher). DNA extraction replicates were run in duplicate for all bacterial populations investigated. Plasmid DNA standard curves were included on each qPCR plate in triplicate and used to convert threshold cycle values to copies per μL of template. The same concentration (12.5 ng) of DNA template and plasmid DNA standard was used in each reaction. Standard curve R square values ranged from 0.990 ± 0.009. Limit of detection was established at 500 copies. The change in bacterial levels were calculated based on logarithms of 16S rRNA gene copy numbers to achieve normal distribution. GraphPad Prism was used for analysis of log transformed data. Statistical significance between time-points was assessed using a two-sided Wilcoxon Signed Rank test with a 95% confidence interval. P ≤ 0.05 was considered statistically significant.
Study of Clostridium species
C. difficile was investigated using two qPCR assays, one targeting the housekeeping gene gluD, and another targeting the 16S gene. Specificity of the primer sets was investigated by standard PCR. DNA template of Clostridium species isolated from gut model samples and identified by MALDI-TOF were amplified using the primers for C. coccoides group, C. leptum group and C. difficile 16S gene (Table 4) to determine primer specificity (Table 1).
Availability of data and materials
The data supporting the conclusions of this article are presented in this published article, and in the additional file 1: Figure S1. Full datasets are available from the corresponding author on reasonable request.
Abbreviations
- CCEY:
-
Cycloserine, cefoxitin egg yolk
- CDI:
-
Clostridioides difficile infection
- cfu:
-
Colony forming unit
- FMT:
-
Faecal microbiota transplantation
- MALDI-TOF:
-
Matrix-Assisted Laser Desorption Ionization-Time of Flight
- N/A:
-
Not applicable
- PBS:
-
Phosphate buffered saline
- qPCR:
-
Quantitative real-time PCR
- rCDI:
-
Recurrent CDI
- rpm:
-
Rotations per minute
- rRNA:
-
Ribosomal ribonucleic acid
- RUs:
-
Relative units
References
Bauer MP, Notermans DW, van Benthem BHB, Brazier JS, Wilcox MH, Rupnik M, et al. Clostridium difficile infection in Europe: a hospital-based survey. Lancet. 2011;377(9759):63–73.
McDonald LC, Gerding DN, Johnson S, Bakken JS, Carroll KC, Coffin SE, et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis. 2018;66:e1–e48.
Eyre DW, Walker AS, Wyllie D, Dingle KE, Griffiths D, Finney J, et al. Predictors of first recurrence of Clostridium difficile infection: implications for initial management. Clin Infect Dis. 2012;55(Suppl 2):S77–87.
Alfayyadh M, Collins DA, Tempone S, McCann R, Armstrong PK, Riley TV, et al. Recurrence of Clostridium difficile infection in the Western Australian population. Epidemiol Infect. 2019;147:e153.
Vardakas KZ, Polyzos KA, Patouni K, Rafailidis PI, Samonis G, Falagas ME. Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: a systematic review of the evidence. Int J Antimicrob Agents. 2012;40(1):1–8.
Kelly CR, Khoruts A, Staley C, Sadowsky MJ, Abd M, Alani M, et al. Effect of fecal microbiota transplantation on recurrence in multiply recurrent Clostridium difficile infection: a randomized trial. Ann Intern Med. 2016;165:609–16.
Cammarota G, Masucci L, Ianiro G, Bibbo S, Dinoi G, Costamagna G, et al. Randomised clinical trial: faecal microbiota transplantation by colonoscopy vs. vancomycin for the treatment of recurrent Clostridium difficile infection. Aliment Pharmacol Ther. 2015;41(9):835–43.
Gough E, Shaikh H, Manges AR. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis. 2011;53(10):994–1002.
Macfarlane GT, Macfarlane S, Gibson GR. Validation of a three-stage compound continuous culture system for investigating the effect of retention time on the ecology and metabolism of Bacteria in the human Colon. Microb Ecol. 1998;35(2):180–7.
Freeman J, O'Neill FJ, Wilcox MH. Effects of cefotaxime and desacetylcefotaxime upon Clostridium difficile proliferation and toxin production in a triple-stage chemostat model of the human gut. J Antimicrob Chemother. 2003;52(1):96–102.
Chilton CH, Freeman J, Crowther GS, Todhunter SL, Wilcox MH. Effectiveness of a short (4 day) course of oritavancin in the treatment of simulated Clostridium difficile infection using a human gut model. J Antimicrob Chemother. 2012;67(10):2434–7.
Chilton CH, Crowther GS, Todhunter SL, Ashwin H, Longshaw CM, Karas A, et al. Efficacy of alternative fidaxomicin dosing regimens for treatment of simulated Clostridium difficile infection in an in vitro human gut model. J Antimicrob Chemother. 2015;70(9):2598–607.
Chilton CH, Crowther GS, Freeman J, Todhunter SL, Nicholson S, Longshaw CM, et al. Successful treatment of simulated Clostridium difficile infection in a human gut model by fidaxomicin first line and after vancomycin or metronidazole failure. J Antimicrob Chemother. 2014;69(2):451–62.
Crowther GS, Chilton CH, Longshaw C, Todhunter SL, Ewin D, Vernon J, et al. Efficacy of vancomycin extended-dosing regimens for treatment of simulated Clostridium difficile infection within an in vitro human gut model. J Antimicrob Chemother. 2016;71(4):986–91.
Moura IB, Buckley AM, Ewin D, Shearman S, Clark E, Wilcox MH, et al. Omadacycline gut microbiome exposure does not induce Clostridium difficile proliferation or toxin production in a model that simulates the proximal, medial, and distal human Colon. Antimicrob Agents Chemother. 2019;63(2):e01581–18.
Chilton CH, Crowther GS, Todhunter SL, Nicholson S, Freeman J, Chesnel L, et al. Efficacy of surotomycin in an in vitro gut model of Clostridium difficile infection. J Antimicrob Chemother. 2014;69(9):2426–33.
Ben-Amor K, Heilig H, Smidt H, Vaughan EE, Abee T, de Vos WM. Genetic diversity of viable, injured, and dead fecal bacteria assessed by fluorescence-activated cell sorting and 16S rRNA gene analysis. Appl Environ Microbiol. 2005;71(8):4679–89.
Rajilic-Stojanovic M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. 2014;38(5):996–1047.
Hayashi. H, Sakamoto. M, Benno. Y. Phylogenetic Analysis of the Human Gut Microbiota Using 16S rDNA Clone Libraries and Strictly Anaerobic Culture-Based Methods. Microbiol Immunol. 2002;46(8):535–48.
Matsuki T, Watanabe K, Fujimoto J, Takada T, Tanaka R. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl Environ Microbiol. 2004;70(12):7220–8.
Louie TJ, Cannon K, Byrne B, Emery J, Ward L, Eyben M, et al. Fidaxomicin preserves the intestinal microbiome during and after treatment of Clostridium difficile infection (CDI) and reduces both toxin reexpression and recurrence of CDI. Clin Infect Dis. 2012;55(Suppl 2):S132–42.
Bartosch S, Fite A, Macfarlane GT, McMurdo ME. Characterization of bacterial communities in feces from healthy elderly volunteers and hospitalized elderly patients by using real-time PCR and effects of antibiotic treatment on the fecal microbiota. Appl Environ Microbiol. 2004;70(6):3575–81.
Rinttila T, Kassinen A, Malinen E, Krogius L, Palva A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol. 2004;97(6):1166–77.
Thorpe CM, Kane AV, Chang J, Tai A, Vickers RJ, Snydman DR. Enhanced preservation of the human intestinal microbiota by ridinilazole, a novel Clostridium difficile-targeting antibacterial, compared to vancomycin. PLoS One. 2018;13(8):e0199810.
Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y. Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol. 2005;54:1093–101.
Mechichi T, Labat M, Patel BK, Woo TH, Thomas P, Garcia JL. Clostridium methoxybenzovorans sp. nov., a new aromatic o-demethylating homoacetogen from an olive mill wastewater treatment digester. Int J Syst Bacteriol. 1999;49(3):1201–9.
Guentzel MN. Escherichia, Klebsiella, Enterobacter, Serratia, Citrobacter, and Proteus. In: Baron S, editor. Medical Microbiology. Galveston (TX): 4th ed: University of Texas Medical Branch at Galveston; 1996.
Dekker JP, Frank KM. Salmonella, Shigella, and yersinia. Clin Lab Med. 2015;35(2):225–46.
Collins MD, Lawson PA, Willems A, Cordoba JJ, Fernandez-Garayzabal J, Garcia P, et al. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int J Syst Bacteriol. 1994;44:812–26.
Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38(7):779–86.
Lawley TD, Croucher NJ, Yu L, Clare S, Sebaihia M, Goulding D, et al. Proteomic and genomic characterization of highly infectious Clostridium difficile 630 spores. J Bacteriol. 2009;191(17):5377–86.
Lawley TD, Walker AW. Intestinal colonization resistance. Immunology. 2013;138(1):1–11.
Oldfield EI, Oldfield EC III, Johnson DA. Clinical update for the diagnosis and treatment of Clostridium difficile infection. World J Gastrointest Pharmacol Ther. 2014;5(1):1–26.
Bernhard AE, Field KG. Identification of nonpoint sources of fecal pollution in coastal waters by using host-specific 16S ribosomal DNA genetic markers from fecal anaerobes. Appl Environ Microbiol. 2000;66(4):1587–94.
Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. 2009;199(8):1177–85.
Delroisse J-M, Boulvin A-L, Parmentier I, Dauphin RD, Vandenbol M, Portetelle D. Quantification of Bifidobacterium spp. and Lactobacillus spp. in rat fecal samples by real-time PCR. Microbiol Res. 2008;163(6):663–70.
Magdesian KG, Leutenegger CM. Real-time PCR and typing of Clostridium difficile isolates colonizing mare-foal pairs. Vet J. 2011;190(1):119–23.
Metcalf D, Sharif S, Weese JS. Evaluation of candidate reference genes in Clostridium difficile for gene expression normalization. Anaerobe. 2010;16(4):439–43.
Acknowledgements
The authors thank Miss Kate Owen and Mrs. Sharie Shearman for the technical assistance, and Cheleste Thorpe for providing the plasmids and scientific support.
Funding
This study was financially supported by the Seres Therapeutics, the RoseTrees Trust (award A1332) and the Leeds Cares. The Funders had no input on project design, data analysis or in the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions to the published study. IBM and AMB designed and performed the experiments and the data analysis. CN, DE and EC performed the gut model experiments. MHW, AMB and CHC provided resources and overseen project design. IBM, MHW, and AMB co-wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The collection/use of faecal donations from healthy adult volunteers following informed verbal consent was approved by the Leeds Institute of Health Sciences and Leeds Institute of Genetics, Health and Therapeutics and Leeds Institute of Molecular Medicine, University of Leeds joint ethics committee (reference HSLTLM/12/061). All volunteers received a participant information document at least 24 h prior to sample collection; this leaflet details the study purpose, why the research is needed and instructions to use the collection kit. When chosen to participate, the volunteers provide the faecal sample to the research team with the understanding that they consented to that sample being used solely for gut model projects. The long-term storage of donor faecal samples for the in vitro gut model experiments was approved by the East Midlands – Leicester South Research Ethics Committee (REC number: 16\EM\0263), IRAS project identification 206,781.
Consent for publication
Not applicable.
Competing interests
IBM has received support to attend meetings from Techlab. AMB has received research funding from Seres, Motif Biosciences, Nabriva, Tetraphase, and Hayashibara Co. CHC has received research funding from Astellas Pharma Europe, Da Volterra and Cubist Pharmaceuticals and support to attend meetings from Astellas. MHW has received honoraria for consultancy work, financial support to attend meetings and research funding from Astellas, AstraZeneca, Abbott, Actelion, Alere, AstraZeneca, Bayer, bioMérieux, Cerexa, Cubist, Da Volterra, Durata, Merck, Nabriva, Pfizer, Qiagen, Roche, Seres, Synthetic Biologics, Summit and The Medicines Company. All other authors: declare.that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Figure S1. Mean gut microbiota populations of C. difficile based on the housekeeping gene gluD in vessel 3 of model A, B and C at the different stages of the experiment. Bar graphs represent the levels in log10 copies/μL measured by qPCR. CD, C. difficile; rCDI, recurrent CDI; FMT, faecal microbiota transplantation. Asterisks represent significant variations by qPCR between time points: *correspond to p < 0.05, and ***correspond to p < 0.0005. (TIF 619 Kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Moura, I.B., Normington, C., Ewin, D. et al. Method comparison for the direct enumeration of bacterial species using a chemostat model of the human colon. BMC Microbiol 20, 2 (2020). https://doi.org/10.1186/s12866-019-1669-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-019-1669-2