
Abstract
Background
G-protein coupled receptors (GPCRs) are the largest family of transmembrane receptors in fungi, where they play important roles in signal transduction. Among them, the Pth11-related GPCRs form a large and divergent protein family, and are only found in fungi in Pezizomycotina. However, the evolutionary process and potential functions of Pth11-related GPCRs remain largely unknown.
Results
Twenty genomes of fungi in Pezizomycotina covering different nutritional strategies were mined for putative Pth11-related GPCRs. Phytopathogens encode much more putative Pth11-related GPCRs than symbionts, saprophytes, or entomopathogens. Based on the phylogenetic tree, these GPCRs can be divided into nine clades, with each clade containing fungi in different taxonomic orders. Instead of fungi from the same order, those fungi with similar nutritional strategies were inclined to share orthologs of putative Pth11-related GPCRs. Most of the CFEM domain-containing Pth11-related GPCRs, which were only included in two clades, were detected in phytopathogens. Furthermore, many putative Pth11-related GPCR genes of phytopathogens were upregulated during invasive plant infection, but downregulated under biotic stress. The expressions of putative Pth11-related GPCR genes of saprophytes and entomopathogens could be affected by nutrient conditions, especially the carbon source. The gene expressions revealed that Pth11-related GPCRs could respond to biotic/abiotic stress and invasive plant infection with different expression patterns.
Conclusion
Our results indicated that the Pth11-related GPCRs existed before the diversification of Pezizomycotina and have been gained and/or lost several times during the evolutionary process. Tandem duplications and trophic variations have been important factors in this evolution.
Similar content being viewed by others

Background
Fungi live in a complex environment, where they receive and integrate abiotic and biotic stimuli, then respond in the manner most appropriate for survival. For example, fungal endophytes in the rhizosphere recognize and colonize specific host plants from which they obtain nutrients [1, 2]. However, the cell wall and membrane, acting as a barrier, separate the interior of the cell from the outside environment [3, 4]. Consequently, communication of cells, both with their environment and with each other, is crucial for the survival of fungi.
Membrane proteins play several essential roles in a cell, including receiving extracellular signals and triggering intracellular responses to them, and the maintenance of interactions between cells [5,6,7]. The fungal cell membrane is equipped with many protein receptors. These receptors sense both abiotic and biotic stimuli from the surrounding environment, and facilitate the response to these stimuli, which may include altering fungal development, morphogenesis, and metabolism [8, 9]. Cell-surface G-protein coupled receptors (GPCRs) are the largest family of transmembrane receptors, and are characterized by seven transmembrane domains anchored in the plasma membrane with an intracellular carboxyl- and extracellular amino-terminus [10, 11]. GPCRs sense a diverse array of stimuli including light, sugars, amino acids, and pheromones [12, 13]. In fungi, many signaling pathways are regulated by GPCRs, such as the mitogen-activated protein kinase and cAMP-dependent protein kinase cascades. These pathways regulate growth, morphogenesis, metabolism, mating, virulence, and stress responses [14, 15].
Many GPCR receptors have been identified in fungi, including pheromone receptors, cAMP receptor-like receptors, carbon-sensing receptors, Stm1-related receptors, and regulator of G protein signaling (RGS) proteins [16]. After first being identified in Saccharomyces cerevisiae [17], Ste2- and Ste3-like pheromone receptors have been found in many ascomycete fungi, while basidiomycete pheromone receptors are only of the Ste3-like type [16, 18]. Neurospora crassa GPR-1 was the first cAMP receptor-like GPCR characterized in ascomycete fungi [19] and the number of this type of GPCR varies among fungal species [16]. S. cerevisiae Gpr1p and N. crassa GPR-4 are carbon-sensing receptors [20, 21], and homologues of Gpr1p and GPR-4 are universally present in fungi [16]. S. pombe Stm1 is involved in the recognition of nitrogen starvation signals [22], and Stm1-related receptors are widely distributed in fungi [16]. RGS proteins are GTPase-activating proteins, which provide negative control of Gα protein signaling [23]. GprK were found to contain an RGS domain in Aspergillus sp. [24], and GprK homologues are present in ascomycetes [16].
A novel class of receptors, the Pth11-related group, was identified by Kulkarni et al. [25]. This group is typified by Magnaporthe oryzae Pth11, a cell-surface integral membrane protein implicated in pathogenesis [25, 26]. These Pth11-related proteins share many characteristics diagnostic of GPCRs, including seven transmembrane regions. For Pth11-related GPCRs, conserved residues (termed as Pth11-domain) occur within the membrane-spanning regions, which is consistent with other GPCRs that sequence conservation is typically limited to the transmembrane sequences [25]. It has been showed that the Pth11-domain is remarkably different from the conserved sequences of other GPCR classes, such as domains conserved in cAMP-, STM1-, and mPR-related GPCRs [25]. The conserved Pth11-domain distinguishes Pth11-related proteins from others and defines a new class of GPCR-like proteins. Except for Pth11-domain, Pth11 also has an amino-terminal extracellular cysteine-rich CFEM domain (pfam05730). However, only a subset of Pth11-related proteins from M. grisea and N. crassa contained the CFEM domain, and these CFEM domain-containing proteins occur together in one clade on the phylogeny tree [25], indicating that the sequences are closely related. The gene duplication may leads to the arisen of these CFEM domain-containing proteins [25].
The Pth11-related GPCRs form a large and diverse protein family [16, 25]. Interestingly, Pth11-related GPCRs were only found in fungi belonging to Pezizomycotina (a subphylum within Ascomycota), while none were found in other subphyla of Ascomycota or Basidiomycota [16, 25]. These results reveal that Pth11-related GPCRs are very ancient in origin, and may have evolved to serve functions specific to this subphylum of fungi. However, the previous studies only focused on some phytopathogens such as M. oryzae and Fusarium graminearum while few of them covered symbionts or entomopathogens. Besides, the evolution of Pth11-related GPCRs and their potential functions are largely unknown, especially at the subphylum level. Recently, an increasing number of genome sequences have become available for fungi in Pezizomycotina, making it possible to mine and compare Pth11-related GPCR sequences. Here we explore the genomes of 20 model organisms in Pezizomycotina with different nutritional strategies and identify Pth11-related GPCRs in the deduced proteomes. The phylogenetic analysis and chromosomal distribution has shed light on the evolution of Pth11-related GPCRs. We also mined expression trends for Pth11-related GPCR genes during growth and invasion, as well as under biotic and abiotic stress, using existing mRNA profiles or microarray datasets.
Results
Identification of putative Pth11-related GPCRs in Pezizomycotina
In total, 20 genomes of fungi in Pezizomycotina were searched for putative Pth11-related GPCRs using a homology (hmmscan and BLAST)-based strategy. These species include members of the Magnaporthales, Ophiostomatales, Sordariales, Glomerellales, Hypocreales, and Eurotiales (Fig. 1), and cover phytopathogens (M. oryzae, Gaeumannomyces graminis, Magnaporthe poae, Verticillium dahlia, Colletotrichum higginsianum, F. graminearum, and Plectospherella cucumerina), symbionts (Harpophora oryzae, and Epichloe festucae), saprophytes (Ophiostoma piceae, N. crassa, Chaetomium globosum, Myceliophthora thermophile, Podospora anserina, Sodiomyces alkalinus, Trichoderma reesei, Aspergillus niger, and Penicillium digitatum), and entomopathogens (Grosmannia clavigera and Metarhizium acridum) (Fig. 1). All the identified proteins were evaluated for the typical topology of seven transmembrane regions, which resulted in 296 putative Pth11-related GPCRs being identified in the 20 proteomes (Fig. 1). The different numbers of predicted Pth11-related GPCRs between the present research and previous studies may result from the newer genome database version and strengthened hmmscan and BLAST cut-off to avoid the false positive.

Phylogenetic relationships and number of putative Pth11-related GPCRs among 20 selected fungal species. S. cerevisiae was used as outgroup. Maximum likelihood (ML) phylogenetic tree shows the evolutionary relationships of 2 Eurotiales species (cyan), 4 Hypocreales species (green), 4 Glomerellales species (blue), 4 Sordariales species (yellow), 2 Ophiostomatales species (purple), and 4 Magnaporthales species (red). The ML bootstrap values are sequentially indicated above the branches. Numbers of Pth11-related GPCRs of the same 20 fungal species are shown on the right of the phylogenetic tree, including 7 phytopathogens, 9 saprophytes, 2 entomopathogens, and 2 symbionts
By multiple sequence alignments of all 296 putative Pth11-related GPCRs, ten blocks of conserved sequences in Pth11-domain were detected (Fig. 2a). Each block contained a conserved motif (Fig. 2b). For examples, motif 1, 2, and 3 were conserved in LXXXR, DD, and GXH/D patterns respectively. The conserved residues of Pth11-domain detected from the 20 proteomes were consistent with previously study in which proteins used were limited to M. grisea and N. crassa [25], indicating the high probability of authenticity of these putative Pth11-related GPCRs.

Multiple sequence alignments and conserved motifs in Pth11-domain. a Alignment of 296 putative Pth11-related GPCRs. Each line refers to one protein sequence. The high conserved residues were in black (yellow in scale bar) while the low conserved ones were in grey (red in scale bar). The ten conserved blocks were indicated above the scale bar. b Sequences of the ten motifs. The ten motifs were detected based on the sequence alignment and each motif respectively associated with a corresponding block
Chromosomal distribution of putative Pth11-related GPCR genes
To determine the chromosomal distribution of putative Pth11-related GPCRs, chromosome maps were constructed for M. oryzae, C. higginsianum, and F. graminearum (Fig. 3 and Additional file 1). These three species all have complete chromosomal (or scaffold) information available, encode more putative Pth11-related GPCRs than others, and belong to different orders. In M. oryzae, the putative Pth11-related GPCR genes are distributed among all seven chromosomes (Fig. 3a). Both chromosome 2 and 3 encoded the highest number (8 genes) of putative Pth11-related GPCR genes, followed by chromosome 6 and 4, encoding 6 and 5 genes respectively. Tandem duplications were found in the chromosome 2 and 6. In C. higginsianum, scaffold 10 was devoid of putative Pth11-related GPCR genes, whereas scaffold 2 encoded the maximum of 5 genes (Fig. 3b). Moreover, tandem duplication was found in scaffold 6. No tandem duplication of putative Pth11-related GPCR genes was found in F. graminearum (Additional file 1).

Chromosomal distribution of putative M. oryzae (a) and C. higginsianum (b) Pth11-related GPCR genes. The chromosome numbers are shown at the top of the chromosomes (M. oryzae) or scaffolds (C. higginsianum), and tandemly duplicated genes are shown in boxes. No putative Pth11-related GPCR genes were detected on scaffold 10 of C. higginsianum
Phylogenetic and hierarchical clustering analysis
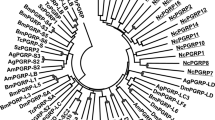
To elucidate the evolutionary relationships among putative Pth11-related GPCRs across fungi in Pezizomycotina, a phylogenetic analysis was performed using the conserved regions of putative Pth11-related GPCR sequences (Fig. 4). Generally, the putative Pth11-related GPCRs from each taxonomic order (i.e., Magnaporthales, Ophiostomatales, Sordariales, Glomerellales, Eurotiales, and Hypocreales) were scattered throughout the phylogenetic tree rather than clustered together. According to the phylogenetic tree, all putative members of the Pth11-related GPCR family can be divided into nine major clades (Fig. 4). Almost all clades were comprised of putative Pth11-related GPCRs from different orders, and no order-specific clade was found. We also performed a hierarchical clustering analysis of the 20 species based on the counts of putative Pth11-related GPCRs in each clade (Fig. 5). This revealed that species with similar nutritional strategies from the same order clustered together, including Magnaporthales (H. oryzae arose from phytopathogens and can be considered a phytopathogen [27]), Sordariales, Eurotiales, and Glomerellales. Meanwhile, species from the same order, but with different nutritional strategies, did not cluster together. To be specific, the phytopathogen F. graminearum (Hypocreales) was clustered with phytopathogens instead of with other fungi in Hypocreales while the saprophyte S. alkalinus (Glomerellales) was clustered with saprophytes rather than other phytopathogens in Glomerellales. Entomopathogens shared a cluster with E. festucae. This may be because E. festucae was derived from insect-parasitic ancestors [28].

Maximum likelihood phylogenetic tree of all predicted Pth11-related GPCRs among 20 selected fungal species. Species belonging to Eurotiales, Hypocreales, Glomerellales, Sordariales, Ophiostomatales, and Magnaporthales are indicated by cyan, green, blue, yellow, purple, and red circles, respectively. The thick red lines denote Pth11-related GPCRs containing a CFEM domain. Bootstrap values greater than 50% are shown at branches

Hierarchical clustering analysis of 20 selected fungal species based on the counts of putative Pth11-related GPCRs in each clade. The clades are defined based on the phylogenetic tree in Fig. 4. Cyan, Eurotiales; Green, Hypocreales; Blue, Glomerellales; Yellow, Sordariales; Purple, Ophiostomatales; Red, Magnaporthales
CFEM domains in putative Pth11-related GPCRs
Only a subset of the identified putative Pth11-related GPCRs (46, 15%) contained cysteine-rich CFEM domain (Fig. 4 and Additional file 2). The P. cucumerina genome encodes the highest number of CFEM-containing Pth11-related GPCRs (8) followed by M. oryzae and C. higginsianum (6). All CFEM-containing Pth11-related GPCRs are included in two clades, i.e., clade 2 (25), and clade 3 (5), except for P. anserina Pa_5_7120 in clade 1, indicating that the sequences are very closely related. Similarly, Kulkarni et al. [25] showed that CFEM-containing Pth11-related GPCRs in M. oryzae and N. crassa occurred in one clade while Gruber et al. [29] showed that this type of GPCR in T. reesei, T. atroviride, and T. virens also clustered together. It is worth noticing that most of CFEM-containing Pth11-related GPCRs were detected in phytopathogens (67%).
Expression patterns of putative Pth11-related GPCR genes in phytopathogens
In order to gain an insight into the possible function of Pth11-related GPCRs, we analyzed the expression patterns of putative Pth11-related GPCR genes under various conditions, including during biotic stress, invasive plant infection, and growth under different nutritional conditions (Fig. 6). Gene expression data for putative Pth11-related GPCRs was mined from publically available datasets, including experiments of GSE65311 [30] and GSE49597 [31] for biotic stress, GSE37886 [32], GSE21908 [33], and GSE33683 [34] for invasive plant infection, and GSE43006 [35], GSE53040 [36], GSE42692 [37], and GSE46155 [38] for nutritional stress. RNA-seq data for H. oryzae were also used to study the invasive infection of plants by H. oryzae [27].

Heatmaps of gene expression of putative Pth11-related GPCRs. a Gene expression in M. oryzae under various treatments. EA105, CHAO, and CHA77 refer to bacteria, including a pseudomonad naturally isolated from rice soil, a P. fluorescens biocontrol strain, and the non-cyanide-producing mutant of CHAO, respectively. 42TEM: heat shock (42 °C for 45 min); MM, minimal media; OS, oxidative stress (treated with methyl viologen); MM-C, carbon limitation; MM-N, nitrogen limitation; Rice: rice at 72 h post-inoculation (hpi); Barley: barley at 72 hpi. b Comparative gene expression of H. oryzae between DAI20 and DAI2 which refer to genes expressed by H. oryzae infecting rice roots at 20 and 2 days after inoculation (DAI), respectively. c Comparative gene expression of C. higginsianum in four stages during the infection process of Arabidopsis. The four stages are: VA (in vitroappressoria), PA (in planta appressoria), BP (biotrophic phase), and NP (necrotrophic phase). d Gene expression of F. graminearum under various treatments. F. graminearum was treated with bacterial MAMPs including flagellin (FLG), lipooligosacharides (LOS), and peptidoglycans (PGN). Time points are 1, 2, and 4 h after treatment with MAMPs. Barley and wheat indicated gene expression during the infection time course (1, 2, and 4 days after inoculation). e Comparative gene expression of T. reesei growing on glucose (Glc), cellulose (Cell), or lactose (Lac) as a sole carbon source. f Comparative gene expression of G. clavigera growing on mannose, olive oil, oleic acid, or terpene as a sole carbon source. g Gene expression of N. crassa under various conditions. N. crassa was subjected to five different nutrient conditions [pectin, orange peel powder (OPP), xylan, avicel, or sucrose as a sole carbon source] and five antifungal compounds [three thioxanthone derivatives (TX129, TX34, TX87), XP13 (a prenylated analogue of 3,4-dihydroxyxanth-9-one,) and D1 (2,4-dihydroxy-3-methylacetophenone)]. Asterisks denote differential expressions greater than twofold change
Expressions of most putative Pth11-related GPCR genes were downregulated when M. oryzae was treated with bacteria that inhibit M. oryzae growth, including EA105 (a pseudomonad naturally isolated from rice soil), CHAO (a Pseudomonas fluorescens biocontrol strain) and CHA77 (the non-cyanide-producing mutant of CHAO) (Fig. 6a). Transcriptional expression was analyzed when F. graminearum was exposed to bacterial MAMPs (microbe-associated molecular patterns), such as flagellin (FLG), lipooligosacharides (LOS), and peptidoglycans (PGN) (Fig. 6d). Three time-points were used (1, 2, and 4 h after treatment with MAMPs). Similarly to the expression patterns in M. oryzae, most putative F. graminearum Pth11-related GPCR genes were downregulated during the biotic stress, especially after 2 h treatment (Fig. 6d). In contrast, upregulation of putative Pth11-related GPCR gene expression was detected during infection of both rice and barley by M. oryzae, including MGG_01884, MGG_09022, MGG_06171, MGG_06535, and MGG_01940 (Fig. 6a). The expressions of putative F. graminearum Pth11-related GPCRs during the infection time course 1, 2, and 4 days after inoculation of plants (wheat and barley) were compared to control (Fig. 6d). Consistent with M. oryzae again, most putative F. graminearum Pth11-related GPCR genes were upregulated during invasive plant infection. Besides, putative Pth11-related GPCR genes could also respond to invasive plant infection for H. oryzae (Fig. 6b) and C. higginsianum (Fig. 6c) by induced regulations with suppressions of a few of them. These results indicated that Pth11-related GPCRs can respond to both biotic stress and invasive plant infection, and in clearly different manners. The two different expression patterns were also suggested by the two separate clusters in the hierarchical clustering analysis (Fig. 6).
Expression patterns of putative Pth11-related GPCR genes in saprophytes and entomopathogens
We examined putative Pth11-related GPCR gene expression in N. crassa treated with five antifungal compounds [three thioxanthone derivatives (TX129, TX34, TX87), XP13 (a prenylated analogue of 3,4-dihydroxyxanth-9-one), and D1 (2,4-dihydroxy-3-methylacetophenone)] and during growth under different nutritional conditions [media with either sucrose, xylan, pectin, orange peel powder (OPP), or avicel as a sole carbon source] (Fig. 6g). Almost all of the putative Pth11-related GPCRs were suppressed by the five antifungal compounds. Downregulation of putative Pth11-related GPCRs was also detected when N. crassa grew under given nutrient conditions. However, NCU02903 and NCU05307 were significantly induced by the 5 nutrient conditions. These two distinct expression patterns were supported by the fact that the GPCRs involved fell into two different clusters (Fig. 6g). Our results not only indicated that putative Pth11-related GPCRs of N. crassa can respond to different nutrient conditions and antifungal compounds, but also showed the clearly opposite expression manners responding to them. We also compared the gene expression of putative Pth11-related GPCR in T. reesei growing on glucose, cellulose, or lactose as carbon sources (Fig. 6e). Induced expression of most putative Pth11-related GPCRs was detected, indicating that a carbon source could affect the expression of putative T. reesei Pth11-related GPCR genes (Fig. 6e). Similar results were found for G. clavigera, which showed that a carbon source, including mannose, olive oil, oleic acid, and terpene, could alter the expression of putative G. clavigera putative genes (Fig. 6f).
Discussion
It has been shown that Pth11 is involved in pathogenesis and is required for the plant pathogen M. oryzae to cause disease [25, 26]. Genomes of phytopathogens such as P. cucumerina, V. dahlia and C. higginsianum in Glomerellales and M. oryzae, G.graminis, and M. poae in Magnaporthales consistently have the largest number of putative Pth11-related GPCRs (from 16 to 41 genes). Compared with phytopathogens, much fewer putative Pth11-related GPCRs were detected in both saprophytes and entomopathogens. The phytopathogen F. graminearum encodes 17 putative pth11-related GPCRs, many more than the other three species (with 7 to 12 each) in Hypocreales. Both H. oryzae and E. festucae are symbionts. However, H. oryzae encodes much more pth11-related GPCRs than E. festucae. One possible explanation is that E. festucae is derived from insect-parasitic ancestors [28], while H. oryzae arose from phytopathogenic ancestors [27]. These results showed that the arsenal of Pth11-related GPCRs might be related to nutritional strategies, especially for phytopathogens. Besides, the hierarchical clustering analysis revealed that species with similar nutritional strategies from the same order clustered together while species from the same order, but with different nutritional strategies were detected in different clusters. These results revealed that instead of fungi from the same order, those fungi with similar nutritional strategies were inclined to share orthologs of putative Pth11-related GPCRs. Moreover, Pth11 has an extracellular amino-terminal CFEM domain [25, 39]. Although only a subset of putative Pth11-related GPCRs contained this fungal-specific cysteine-rich CFEM domain, most of CFEM-containing Pth11-related GPCRs were detected in phytopathogens and were very closely related. This phenomenon also indicated that trophic variations have been important factors in the evolution of Pth11-related GPCRs. Furthermore, the topology of the phylogenetic tree indicated that each order’s putative Pth11-related GPCRs were derived from GPCRs of their common ancestors and that Pth11-related GPCRs gained and/or lost several times during the evolutionary process. Overall, these results make it fairly safe to infer that Pth11-related GPCRs existed before the divergence of Pezizomycotina, and later evolved independently in a species-specific manner. And during the evolution of Pth11-related GPCRs, the different nutritional strategies of these fungi could be an important evolutionary stress.
The evolution of Pth11-related GPCRs involved in trophic variations of fungi in Pezizomycotina could also be revealed by their possible functions. We analyzed the expression pattern of putative Pth11-related GPCR genes under various conditions, including during biotic stress, invasive plant infection, and growth under different nutritional conditions. Expressions of most putative Pth11-related GPCR genes from both M. oryzae and F. graminearum were downregulated during the biotic stress while upregulation were detected during invasive plant infection by both of them. The two clearly different expression patterns revealed that Pth11-related GPCRs can respond to both biotic stress and invasive plant infection for phytopathogens. For saprophytes, almost all the putative Pth11-related GPCRs from N. crassa were suppressed by the antifungal compounds but some of them were induced when N. crassa were subjected to different nutrient conditions. Similar results were also found by Cabrera et al. [9], who revealed that many Pth11-related GPCRs are related to chemical sensitivity or nutritional phenotypes by analyzing the phenotypes of mutants. We also found that carbon source could affect the expression of putative Pth11-related GPCR genes of T. reesei and G. clavigera. The results revealed the common functions of Pth11-related GPCRs in respond to nutritional conditions for saprophytes and entomopathogens.
Expression patterns of putative Pth11-related GPCR genes in M. oryzae were also detected under abiotic stresses including growth on minimal medium, carbon and nitrogen starvation, heat shock (42 °C for 45 min), and oxidative stress (treated with methyl viologen). Interestingly, the hierarchical clustering analysis showed that the abiotic stress and invasive plant infection clustered together, indicating the similarity in expression patterns of putative Pth11-related GPCR genes between abiotic stress and invasive plant infection. These similar expression patterns may have resulted from that M. oryzae typically encountering nutrient-limited environments at the invasive growth stage [33].
The key role of tandem duplication in the evolution of other gene families has been reported, including the P450 family [40]. It has also been shown that gene duplication often plays a central role in both fungal and plant diversification, and is a key process generating the raw material necessary for adaptive evolution [41, 42]. Tandem duplications were detected in the chromosomes of both M. oryzae and C. higginsianum, indicating that tandem duplication may contribute to the evolution of the Pth11-related GPCR family. However, no tandem duplication of putative Pth11-related GPCR genes was found in F. graminearum (Additional file 1), indicating the complex evolutionary history of Pth11-related GPCRs.
Conclusions
This study provided a thorough examination of 20 genomes in Pezizomycotina for putative Pth11-related GPCRs. The 20 selected fungi cover different nutritional strategies, including phytopathogens, symbionts, saprophytes, and entomopathogens. To elucidate the evolution of putative Pth11-related GPCRs, we performed a chromosome distribution and phylogenetic analysis of them. This analysis indicated that putative Pth11-related GPCRs existed before the divergence of Pezizomycotina, and that the GPCRs in each species were derived from GPCRs of their common ancestors. During this evolutionary process, putative Pth11-related GPCRs could have been gained and lost several times, possibly involving tandem duplication. Our results showed that putative Pth11-related GPCRs could respond to bacterial challenges, antifungal compounds, different nutritional conditions, and invasive plant infection, and different expression patterns were used to in response to these stimuli. Based on the common functions of putative Pth11-related GPCRs in respond to nutritional conditions and the results of fungi with similar nutritional strategies were inclined to share orthologs of putative Pth11-related GPCRs, we suggested that the different nutritional strategies of fungi could have been an important evolutionary stress during the evolution of Pth11-related GPCRs. It is worth mentioning that the proteins identified as putative Pth11-related GPCRs in this study have only been characterized in silico. Compared with only three types of G protein in most fungi, a large number of putative Pth11-related GPCRs were detected. Therefore, determining the intracellular interactions of Pth11-related GPCRs and their signaling output will help us understand how fungi adapt to different challenges and nutritional conditions.
Methods
Identification of putative Pth11-related GPCRs
Twenty genome sequences and deduced proteomes of Pezizomycotina were used from the following fungi: M. oryzae, G. graminis, M. poae [43], H. oryzae [44], G. clavigera [45], C. globosum [46], N. crassa [47], V. dahlia [48], C. higginsianum [49], F. graminearum [50], E. festucae [51], M. acridum [52], T. reesei [53], P. digitatum [54], A. niger [55], S. alkalinus [56], P. cucumerina [57], P. anserine [58], M. thermophile [59], and O. piceae [60]. Genome sequence of S. cerevisiae [61] was used as outgroup. These species cover Magnaporthales, Ophiostomatales, Sordariales, Glomerellales, and Hypocreales, and comprise phytopathogens (M. oryzae, G. graminis, M. poae, V. dahlia, C. higginsianum, F. graminearum and P. cucumerina), symbionts (H. oryzae, and E. festucae), saprophytes (O. piceae, N. crassa, C. globosum, M. thermophile, P. anserina, S. alkalinus, T. reesei, A. niger, and P. digitatum), and entomopathogens (G. clavigera and M. acridum). A pipeline was used to identify the putative Pth11-related GPCRs in the 21 selected proteomes. Firstly, as the Pth11-domain distinguishes Pth11-related proteins from other class of GPCR-like proteins [25], the hmmscan program from the HMMER v3.1 package [62] was used to search for the Pth11-domain across all the 21 proteomes with an e-value cutoff of 1e-20. Then the obtained Pth11-domain containing proteins were used in a BLASTP search against Pth11-related GPCRs of M. oryzae [25]. An e-value limit of 1e-09 was applied, and all proteins that had at least 30% identity and 80% overlap over the length of the proteins were retained [25]. Finally, the obtained proteins were evaluated for the typical seven-transmembrane domain by TMHMM, HMMTOP, and Phobius [63,64,65], and proteins with seven or more transmembrane domains predicted by at least two algorithms were retained as putative Pth11-related GPCRs and used for further analysis. By this analysis pipeline, the authenticity of these predicted Pth11-related GPCRs was highly improved.
Chromosomal organization of putative Pth11-related GPCRs
Chromosome location images were generated using MapInspect software to localize putative Pht11-related GPCRs of M. oryzae, C. higginsianum, and F. graminearum. Any putative Pth11-related GPCRs separated by no more than 15 genes were identified as tandemly duplicated genes.
Protein alignments and phylogenetic analysis
Protein sequences were aligned using ClustalW v2 [66]. To select conserved regions, the alignments were analyzed with Gblocks v0.91b [67] using the default parameters. The best amino acid substitution model was chosen using ProtTest v3.2 [68], and LG + G + F model was selected as the best-fit model for the datasets. The phylogenetic tree of putative Pth11-related GPCRs was constructed in MEGA v7 [69] using maximum likelihood (ML) methods with the best-fit model followed by bootstrap analysis (1000 bootstrap replications). To infer phylogenetic relationships among the 20 species, 100 clusters of 1:1 orthologs were chosen based on our previous study [27]. The proteins of the 100 orthologs were aligned and then concatenated. Phylogenetic analyses were performed using the ML criterion implemented in RAxML [70] through the RAxML-HPC BlackBox web server at the Cyber Infrastructure for Phylogenetic Research with LG + G + I model. The sequence logo was created using WebLogo v2.8.2 [71] based on a multiple sequence alignment of 296 putative Pth11-related GPCRs.
CFEM domain search
The CFEM domain architectures of putative Pth11-related GPCRs were predicted using two search methods, including Pfam [72], and Conserved Domain Search (CDSearch) [73] applying the default settings for each.
Expression analysis
Expression data were mined for putative Pth11-related GPCR genes from several datasets, including RNA sequencing and microarray data, which were downloaded from the Gene Expression Omnibus. The datasets with accession numbers GSE65311, GSE37886 (F. graminearum), GSE21908, GSE49597 (M. oryzae), GSE33683 (C. higginsianum), GSE43006 (G. clavigera), GSE53040, GSE42692 (N. crassa), and GSE46155 (T. reesei) were analyzed. For H. oryzae, RNA-seq data from a previous study [27] were used. These data pertained to different environmental conditions, including biotic and abiotic stress conditions and various stages of colony development. The data were visualized using heatmaps generated with the heatmap.2 package in R, which is based on log2 fold changes after normalization.
Abbreviations
- BLAST:
-
Basic local alignment search tool
- GPCR:
-
G-protein coupled receptors
- GPCRDB:
-
GPCR database
- MAMP:
-
Microbe-associated molecular patterns
References
Rodriguez RJ, White JF, Arnold AE, Redman RS. Fungal endophytes: diversity and functional roles. New Phytol. 2009;182:314–30.
Xu XH, Wang C, Li SX, Su ZZ, Zhou HN, Mao LJ, et al. Friend or foe: differential responses of rice to invasion by mutualistic or pathogenic fungi revealed by RNAseq and metabolite profiling. Sci Rep. 2015;5:13624.
Robertson JD. The ultrastructure of cell membranes and their derivatives. Biochem Soc Symp. 1959;16:3–43.
Zimmermann W, Rosselet A. Function of the outer membrane of Escherichia Coli as a permeability barrier to beta-lactam antibiotics. Antimicrob Agents Chemother. 1977;12:368–72.
von Heijne G. Membrane-protein topology. Nat Rev Mol Cell Biol. 2006;7:909–18.
Engel A, Gaub HE. Structure and mechanics of membrane proteins. Annu Rev Biochem. 2008;77:127–48.
Müller DJ, Wu N, Palczewski K. Vertebrate membrane proteins: structure, function, and insights from biophysical approaches. Pharmacol Rev. 2008;60:43–78.
Elion EA. Pheromone response, mating and cell biology. Curr Opin Microbiol. 2000;3:573–81.
Cabrera IE, Pacentine IV, Lim A, Guerrero N, Krystofova S, Li L, et al. Global analysis of predicted G protein-coupled receptor genes in the filamentous fungus, Neurospora crassa. G3 (Bethesda). 2015;5:2729–43.
Shukla AK, Singh G, Ghosh E. Emerging structural insights into biased GPCR signaling. Trends Biochem Sci. 2014;39:594–602.
Chini B, Parenti M, Poyner DR, Wheatley M. G-protein-coupled receptors: from structural insights to functional mechanisms. Biochem Soc Trans Biochem Soc Trans. 2013;41:135–6.
Maller JL. Fishing at the cell surface. Science. 2003;300:594–5.
Hamm HE. The many faces of G protein signaling. J Biol Chem. 1998;273:669–72.
Lengeler KB, Davidson RC, D’souza C, Harashima T, Shen WC, Wang P, et al. Signal transduction cascades regulating fungal development and virulence. Microbiol Mol Biol Rev. 2000;64:746–85.
Rispail N, Soanes DM, Ant C, Czajkowski R, Grünler A, Huguet R, et al. Comparative genomics of MAP kinase and calcium-calcineurin signalling components in plant and human pathogenic fungi. Fungal Genet Biol. 2009;46:287–98.
Li L, Wright SJ, Krystofova S, Park G, Borkovich KA. Heterotrimeric G protein signaling in filamentous fungi. Annu Rev Microbiol. 2007;61:423–52.
Dohlman HG, Thorner JW. Regulation of G protein-initiated signal transduction in yeast: paradigms and principles. Annu Rev Biochem. 2001;70:703–54.
Casselton LA, Olesnicky NS. Molecular genetics of mating recognition in basidiomycete fungi. Microbiol Mol Biol Rev. 1998;62:55–70.
Krystofova S, Borkovich KA. The predicted G-protein-coupled receptor GPR-1 is required for female sexual development in the multicellular fungus Neurospora crassa. Eukaryot Cell. 2006;5:1503–16.
Lorenz MC, Pan X, Harashima T, Cardenas ME, Xue Y, Hirsch JP, et al. The G protein-coupled receptor Gpr1 is a nutrient sensor that regulates pseudohyphal differentiation in Saccharomyces Cerevisiae. Genetics. 2000;154:609–22.
Li L, Borkovich KA. GPR-4 is a predicted G-protein-coupled receptor required for carbon source-dependent asexual growth and development in Neurospora crassa. Eukaryot Cell. 2006;5:1287–300.
Chung KS, Won M, Lee SB, Jang YJ, Hoe KL, Kim DU, et al. Isolation of a novel Gene from Schizosaccharomyces pombe: Stm1 encoding a seven-transmembrane loop protein that may couple with the Heterotrimeric Gα2 protein, Gpa2. J Biol Chem. 2001;276:40190–201.
Chen JG, Willard FS, Huang J, Liang J, Chasse SA, Jones AM, et al. A seven-transmembrane RGS protein that modulates plant cell proliferation. Science. 2003;301:1728–31.
Lafon A, Han KH, Seo JA, Yu JH, d’Enfert C. G-protein and cAMP-mediated signaling in aspergilli: a genomic perspective. Fungal Genet Biol. 2006;43:490–502.
Kulkarni RD, Thon MR, Pan H, Dean RA. Novel G-protein-coupled receptor-like proteins in the plant pathogenic fungus Magnaporthe grisea. Genome Biol. 2005;6:R24.
DeZwaan TM. Magnaporthe grisea Pth11p is a novel plasma membrane protein that mediates Appressorium differentiation in response to inductive substrate cues. Plant Cell. 1999;11:2013–30.
Xu XH, Su ZZ, Wang C, Kubicek CP, Feng XX, Mao LJ, et al. The rice endophyte Harpophora oryzae genome reveals evolution from a pathogen to a mutualistic endophyte. Sci Rep. 2014;4:5783.
Spatafora JW, Sung GH, Sung JM, Hywel-Jones NL, White JF. Phylogenetic evidence for an animal pathogen origin of ergot and the grass endophytes. Mol Ecol. 2007;16:1701–11.
Gruber S, Omann M, Zeilinger S. Comparative analysis of the repertoire of G protein-coupled receptors of three species of the fungal genus Trichoderma. BMC Microbiol. 2013;13:108.
Ipcho S, Sundelin T, Erbs G, Kistler HC, Newman M-A, Olsson S. Fungal innate immunity induced by bacterial microbe-associated molecular patterns (MAMPs). G3 (Bethesda). 2016;6:1585–95.
Spence CA, Raman V, Donofrio NM, Bais HP. Global gene expression in rice blast pathogen Magnaporthe oryzae treated with a natural rice soil isolate. Planta. 2014;239:171–85.
Harris LJ, Balcerzak M, Johnston A, Schneiderman D, Ouellet T. Host-preferential Fusarium graminearum gene expression during infection of wheat, barley, and maize. Fungal Biol. 2016;120:111–23.
Mathioni SM, Belo A, Rizzo CJ, Dean RA, Donofrio NM. Transcriptome profiling of the rice blast fungus during invasive plant infection and in vitro stresses. BMC Genomics. 2011;12:20.
O’Connell RJ, Thon MR, Hacquard S, Amyotte SG, Kleemann J, Torres MF, et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat Genet. 2012;44:1060–5.
Wang Y, Lim L, Madilao L, Lah L, Bohlmann J, Breuil C. Gene discovery for enzymes involved in limonene modification or utilization by the mountain pine beetle-associated pathogen Grosmannia clavigera. Appl Environ Microbiol. 2014;80:4566–76.
Pedro Gonçalves A, Silva N, Oliveira C, Kowbel DJ, Glass NL, Kijjoa A, et al. Transcription profiling of the Neurospora crassa response to a group of synthetic (thio)xanthones and a natural acetophenone. Genomics Data. 2015;4:26–32.
Benz PJ, Chau BH, Zheng D, Bauer S, Glass NL, Somerville CR. A comparative systems analysis of polysaccharide-elicited responses in Neurospora crassa reveals carbon source-specific cellular adaptations. Mol Microbiol. 2014;91:275–99.
Bischof R, Fourtis L, Limbeck A, Gamauf C, Seiboth B, Kubicek CP. Comparative analysis of the Trichoderma Reesei transcriptome during growth on the cellulase inducing substrates wheat straw and lactose. Biotechnol Biofuels. 2013;6:127.
Kulkarni RD, Kelkar HS, Dean RA. An eight-cysteine-containing CFEM domain unique to a group of fungal membrane proteins. Trends Biochem Sci. 2003;28:118–21.
Doddapaneni H, Chakraborty R, Yadav JS. Genome-wide structural and evolutionary analysis of the P450 monooxygenase genes (P450ome) in the white rot fungus Phanerochaete chrysosporium: evidence for gene duplications and extensive gene clustering. BMC Genomics. 2005;6:92.
Flagel LE, Wendel JF. Gene duplication and evolutionary novelty in plants. New Phytol. 2009;183:557–64.
Wapinski I, Pfeffer A, Friedman N, Regev A. Natural history and evolutionary principles of gene duplication in fungi. Nature. 2007;449:54–61.
The Magnaporthe comparative genome database. https://www.broadinstitute.org/scientificcommunity/science/projects/fungal-genome-initiative/magnaporthe-comparative-genomics-proj. Accessed 16 Mar 2016.
The Harpophora oryzae genome database. http://www.ncbi.nlm.nih.gov/nuccore/667821394. Accessed 16 Mar 2016.
The Grosmannia clavigera genome database. http://genome.jgi.doe.gov/Grocl1/Grocl1.home.html. Accessed 16 Mar 2016.
The Chaetomium globosum genome database. http://genome.jgi.doe.gov/Chagl_1/Chagl_1.home.html. Accessed 16 Mar 2016.
The Neurospora crassa genome database. http://genome.jgi.doe.gov/Neucr_trp3_1/Neucr_trp3_1.home.html. Accessed 16 Mar 2016.
The Verticillium dahlia genome database. http://genome.jgi.doe.gov/Verda1/Verda1.home.html. Accessed 16 Mar 2016.
The Colletotrichum higginsianum genome database. http://genome.jgi.doe.gov/Colhi1/Colhi1.home.html. Accessed 16 Mar 2016.
The Fusarium graminearum genome database. http://genome.jgi.doe.gov/Fusgr1/Fusgr1.home.html. Accessed 16 Mar 2016.
The Epichloe festucae genome database. http://csbio-l.csr.uky.edu/ef894. Accessed 16 Mar 2016.
The Metarhizium acridum genome database. http://genome.jgi.doe.gov/Metac1/Metac1.home.html. Accessed 16 Mar 2016.
The Trichoderma reesei genome database. http://genome.jgi.doe.gov/Trire2/Trire2.home.html. Accessed 16 Mar 2016.
The Penicillium digitatum genome database. http://genome.jgi.doe.gov/Pendi1/Pendi1.home.html. Accessed 10 June 2017.
The Aspergillus niger genome database. http://genome.jgi.doe.gov/Aspni_DSM_1/Aspni_DSM_1.home.html. Accessed 10 June 2017.
The Sodiomyces alkalinus genome database. http://genome.jgi.doe.gov/Sodal1/Sodal1.home.html. Accessed 10 June 2017.
The Plectospherella cucumerina genome database. http://genome.jgi.doe.gov/Plecu1/Plecu1.home.html. Accessed 10 June 2017.
The Podospora anserine genome database. http://genome.jgi.doe.gov/Podan2/Podan2.home.html. Accessed 10 June 2017.
The Myceliophthora thermophile genome database. http://genome.jgi.doe.gov/Spoth2/Spoth2.home.html. Accessed 10 June 2017.
The Ophiostoma piceae genome database. http://genome.jgi.doe.gov/Ophpic1/Ophpic1.home.html. Accessed 10 June 2017.
The Saccharomyces cerevisiae database. http://genome.jgi.doe.gov/Sacce1/Sacce1.home.html. Accessed 10 June 2017.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server issue):W29–37.
Tusnády GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849–50.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80.
Käll L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338:1027–36.
Larkin MA, Blackshields G, Brown NP, Chenna R, Mcgettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8.
Castresana J. Selection of conserved blocks from multiple alignments for their use in Phylogenetic analysis. Mol Biol Evol. 2000;17:540–52.
Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–5.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90.
Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–90.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–30.
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43:D222–6.
Acknowledgements
Not applicable.
Funding
The research was funded by the National Key Research and Development Program of China (2016YFD0800803), the National Natural Science Foundation of China (31501689 and 31400328), the Science Foundation of Jiangsu Province, China (BK20150670 and BK20140697), and the Fundamental Research Funds for the Central Universities (KJQN201641).
Availability of data and materials
The expression data of this article are available in Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) in NCBI under accession numbers:
GSE65311, GSE37886, GSE21908, GSE49597, GSE33683, GSE43006, GSE53040, GSE42692, and GSE46155.
Author information
Authors and Affiliations
Contributions
XX and CC conceived and designed the experiments; XX and GL performed the experiments; XX, GL, LL, and ZS analyzed the data; XX and CC wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All authors are aware of the publication.
Consent for publication
All authors consent to the submitted version of this manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Chromosomal distribution of putative F. graminearum Pth11-related GPCR genes. Chromosome numbers are shown at the top of the chromosomes. (TIFF 39 kb)
Additional file 2:
Predicted Pth11-related GPCRs among thirteen fungi belonging to Pezizomycotina. (XLSX 24 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xu, X., Li, G., Li, L. et al. Genome-wide comparative analysis of putative Pth11-related G protein-coupled receptors in fungi belonging to Pezizomycotina. BMC Microbiol 17, 166 (2017). https://doi.org/10.1186/s12866-017-1076-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-017-1076-5