
Abstract
Background
Characterizing microbial communities via next-generation sequencing is subject to a number of pitfalls involving sample processing. The observed community composition can be a severe distortion of the quantities of bacteria actually present in the microbiome, hampering analysis and threatening the validity of conclusions from metagenomic studies. We introduce an experimental protocol using mock communities for quantifying and characterizing bias introduced in the sample processing pipeline. We used 80 bacterial mock communities comprised of prescribed proportions of cells from seven vaginally-relevant bacterial strains to assess the bias introduced in the sample processing pipeline. We created two additional sets of 80 mock communities by mixing prescribed quantities of DNA and PCR product to quantify the relative contribution to bias of (1) DNA extraction, (2) PCR amplification, and (3) sequencing and taxonomic classification for particular choices of protocols for each step. We developed models to predict the “true” composition of environmental samples based on the observed proportions, and applied them to a set of clinical vaginal samples from a single subject during four visits.
Results
We observed that using different DNA extraction kits can produce dramatically different results but bias is introduced regardless of the choice of kit. We observed error rates from bias of over 85% in some samples, while technical variation was very low at less than 5% for most bacteria. The effects of DNA extraction and PCR amplification for our protocols were much larger than those due to sequencing and classification. The processing steps affected different bacteria in different ways, resulting in amplified and suppressed observed proportions of a community. When predictive models were applied to clinical samples from a subject, the predicted microbiome profiles were better reflections of the physiology and diagnosis of the subject at the visits than the observed community compositions.
Conclusions
Bias in 16S studies due to DNA extraction and PCR amplification will continue to require attention despite further advances in sequencing technology. Analysis of mock communities can help assess bias and facilitate the interpretation of results from environmental samples.
Similar content being viewed by others


Background
Next-generation sequencing technology (NGS) allows a much deeper characterization of the structure of microbial communities using metagenomic approaches. Metagenomic surveys often use a hypervariable region of the highly-conserved and universal 16S rRNA gene as a phylogenetic marker. Bias introduced in the processing steps of such surveys masks the true community composition so that there are large discrepancies in the proportion of gram negative bacteria observed using next generation sequencing, microscopy, and culture-based methods [1]. An objective of microbiome experiments is to characterize the community composition, including the relative quantities of species in sampled environments. An accurate depiction of microbial community composition via next generation sequencing requires a careful consideration of bias introduced during sample processing [2,3].
Many sources of bias have been identified in 16S rRNA studies using NGS including PCR amplification [4-11], DNA extraction protocol [5,12,13], sequencing artifacts [8,14-18], DNA copy number [19], sampling depth [7,20,21], and primer design [22-25]. Previous studies typically isolate one or two sources of bias, suggest experimental practices that mitigate the effects, and acknowledge that other sources of bias remain. Examples of recommendations for mitigating bias include performing triple DNA extraction [12], using multiple combinations of DNA extraction and PCR amplification protocols [5], and reducing the number of PCR cycles to avoid chimera formation [6].
Few studies have attempted to create models for neutralizing bias in environmental samples. One exception is a strategy proposed for counteracting the portion of bias due to differences in DNA copy number among bacteria [19]. The method applies a phylogenetic and ancestral state placement of sample sequences among a reference database of bacteria with known 16S copy numbers.
We perceive the need for three kinds of quality control in microbiome experiments. The first is the need to monitor batch effects of different sample processing runs. The use of identical mock (or even environmental) samples as positive controls and sequencing pure PCR product as a negative control can help to identify problems with batches and drift. The second type of quality control is based on the variation produced by the choice of sample processing protocols. The same sample processed at labs that use different protocols can produce different results. The Microbiome Quality Control project [3] is studying the effects of different choices in protocols and seeks to understand which choices contribute the most to variation. The third type of quality control is understanding the difference between observed and actual community compositions for particular choices of protocols for a lab. Depending on the environment of interest, labs will engineer their procedures so that they are sure to detect organisms of interest for the particular environment. Understanding the bias resulting from these choices of protocols is important because no matter what choice is made, bias will remain. The experiment reported here was designed to understand the magnitude and nature of bias introduced by a particular choice of protocols.
The methodology proposed here represents, to our knowledge, the first attempt to (1) create comprehensive models for predicting community composition in environmental samples based on observed proportions, (2) quantify the contribution of bias of different sample processing steps in 16S experiments, and (3) identify statistically significant relationships between bacterial signals. Additional distinguishing features of this study include the deep sequencing employed and the species-level taxonomic classification of reads.
This paper proposes a set of mixture experiments involving small “mock” communities, artificial microbial communities created by mixing known quantities of bacterial isolates, DNA clones, or PCR product. Mock communities are often used for ground-truthing and quantifying bias [4,9]. While mixture experiments occur most frequently in areas such as chemistry and agriculture, they have also been applied in the biological sciences. Mixture designs can be used for screening complex medium components in the cultivation of bacteria and evaluating the influence of nutrients on bacterial byproducts and growth [26-30]. For instance, Kiviharju et al. [26] apply a mixture design for the screening of suitable complex medium components in the cultivation of S. peucetius var. caesius, an aerobic bacterium that produces doxorubicin as a secondary metabolite. Rispoli and Shah [27] use mixture experiments to evaluate the influence of six nutrient elements on production of cutinase from the fungus Colletotrichum lindemuthianum. For other examples, see [28-30].
We report on an application of the proposed experimental protocol to an analysis of seven vaginally-relevant bacteria and apply the results to clinical samples.
Results and discussion
Different DNA extraction kits introduced different bias
We analyzed a single mock community and varied the DNA extraction kit and the number of PCR cycles. The mock community consists of 21 bacterial/archaeal strains from 18 genera [31] that are not necessarily associated with the human vagina. Taxonomic classification was performed using the RDP classifier [32] (see Methods). The choice of DNA extraction kit led to the most striking differences between the protocols tested (Additional file 1). Relative to the Powersoil kit, using the Qiagen kit increased the observed proportion of Enterococcus by about 50% while suppressing the observed proportions of Neisseria, Bacillus, Pseudomonas, and Porphyromonas. In contrast, changing the number of PCR cycles from 30 to 35 affected the observed proportions only slightly. The small changes due to PCR cycle number agree with previous studies that showed that while chimera formation increases with cycle number, the observed community structure is retained [6,9,10,33,34].
Each combination of extraction kit and cycle number produced results that were dramatically different from the actual mixing proportions. The differences between the observed and actual proportions were different for the different extraction kits. The results for each of the samples produced underestimates of Lactobacillus (the only species included in the mock community was L. gasseri) and Streptococcus (the mock community included S. pneumoniae, S. mutans and S. agalactiae).
Mixture experiments and mixture effect models for quantifying and characterizing bias in 16S metagenomic studies
A mixture design is an experiment in which a response of interest is assumed to depend only on the relative proportions of the components present in the mixture. If the response changes when the proportions of those components making up the mixture are altered, then the response is said to be a measure of the joint blending property of the components of the mixture [35]. The distinguishing feature of mixture experiments is that the mixture components are subject to a constraint requiring that the proportions sum to one. Due to this constraint on the mixture components, non-standard statistical models are required to model the response. Mixture effect models [36] allow for prediction of the response for given proportions of mixture components as well as evaluation of relationships among the components.
We developed the following protocol for quantifying and characterizing bias in 16S metagenomic studies:
-
1.
Decide upon a small subset of bacteria whose measurement is of interest. We selected seven vaginally-relevant species based on their prevalence in clinical samples, suspected importance in disease mechanisms, and ability to be cultured.
-
2.
Based on the number of bacteria selected and the number of runs available, generate an experimental design. The 80-run mixture experiment for our application was a D-optimal design [37] containing 65 unique treatment combinations and 15 replicate samples. Replicate runs were used to obtain an estimate of the pure error variance.
The D-optimal design that we used requires at least 63 runs:
$$\left(7 \atop 3\right) + \left(7 \atop 2\right) + 7 = 63, $$in order to fit a special cubic model (see Methods). The 17 additional runs in the design included two other unique treatment combination for testing lack of fit and 15 replicates. With n bacteria, the same design would require the number obtained by replacing 7 with n in the formula above. For example, an analogous model for 12 bacteria would require a minimum of 298 runs.
-
3.
Randomize the design for three mixture experiments. The treatment combinations and placement on plates were randomized to alleviate effects of bias due to experimental conditions. Each row of the experimental design in Additional file 2 contains a treatment combination that prescribes the proportion of cells, DNA, or PCR product from each strain of bacteria used in the construction of a mock community.
-
4.
Prepare and process mock communities according to the experimental design. Preparing mock communities for each experiment is described below and illustrated in Figure 1.
-
Experiment 1. Create mock communities by mixing prescribed quantities of cells from each organism. Grow each isolate to exponential phase and determine cell density through estimates of viable cell counts and optical density; the combined approach improves the accuracy of estimates. Combine bacteria to form mock communities and subject the samples to DNA extraction, PCR amplification, sequencing, and taxonomic classification.
Figure 1 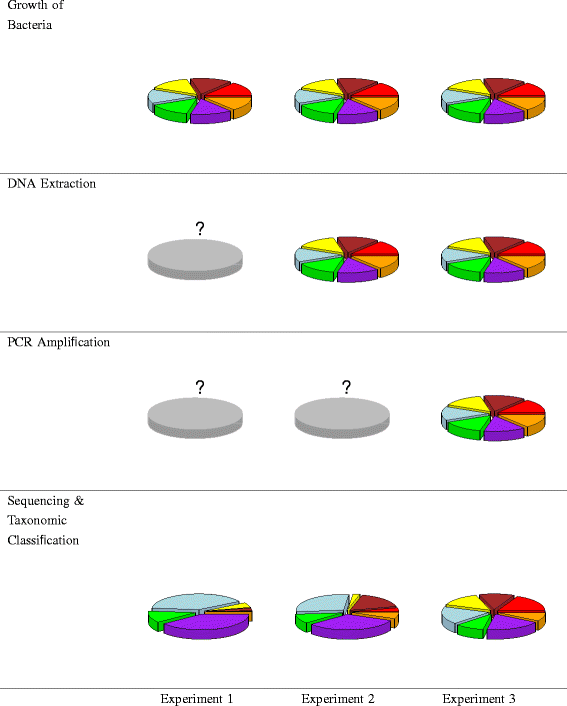
Schematic of three mixture experiments and observed results. In Experiment 1, bacterial cultures were mixed so that communities were comprised of equal numbers of cells. In Experiment 2, DNA was extracted from pure bacterial cultures and then mixed so that communities were comprised of equal amounts of DNA. In Experiment 3, DNA was extracted from pure bacterial cultures and subjected to PCR and PCR product was mixed so that communities are comprised of equal amounts of PCR product. The pie charts in the bottom row are the observed results for a sample that consisted of equal proportions of seven bacteria for each experiment. The pie charts in the other rows represent the prescribed mixing ratios (each slice is of equal size). Key: red - G. vaginalis, orange - S. agalactiae, purple - S. amnii,green - P. bivia, light blue - L. iners, yellow - L. crispatus, brown - A. vaginae.
-
Experiment 2. Create mock communities by mixing proportions of gDNA. Extract gDNA from pure cultures of each bacterial strain. Measure DNA concentration and mix in the proportions described by the experimental design. Then process each sample by PCR amplification, sequencing, and taxonomic classification.
-
Experiment 3. Create mock communities by mixing equal proportions of PCR product. Begin by extracting gDNA from the pure cultures of each bacterial species. Subject the pure gDNA to PCR amplification. Mix the PCR products according to the experimental design. Sequence each sample and classify the reads.
-
-
5.
Compare the differences in the results of each experiment. Comparing the results of Experiment 1 with the prescribed mixing ratios gives a measurement of the total bias. If x i is the prescribed mixing ratio for bacterium i and \(\hat {x}_{i}\) is the observed proportion, then the bias is the difference \(\hat {x}_{i} - x_{i}\). A negative value indicates that the bacterial signal is suppressed, while a positive value indicates that the signal is amplified.
Comparing the results of Experiments 1 and 2 isolates the effects of the DNA extraction protocol. Similarly, comparing the results of Experiments 2 and 3 isolates the effects of bias due to the PCR amplification protocol. Comparing the results of Experiment 3 with the prescribed mixing ratios isolates the effects of sequencing and taxonomic classification. The pie charts at the bottom of Figure 1 show the results for a sample that contained equal proportions of the seven bacteria for each of our experiments. Similar information would be generated for each mock community in each experiment, facilitating analysis via modeling.
-
6.
Fit mixture effect models to regress the observed proportions of reads against functions of the prescribed mixing ratios. Significant terms in the models can reveal pairs or groups of bacteria that, when present together in a sample, amplify or suppress the observed proportions of other bacteria.
-
7.
Fit models to regress the prescribed mixing ratios against the observed proportions of reads for prediction of actual community composition based on the observed community composition. Use cross-validation to estimate accuracy for clinical/environmental samples.

In the sections that follow, we report on the results of our application of this protocol for seven species: Atopobium vaginae, Gardnerella vaginalis, Lactobacillus crispatus, Lactobacillus iners, Prevotella bivia, Sneathia amnii, and Streptococcus agalactiae.
Contaminating bacteria did not significantly contribute to bias in small mock communities
A total of 3.9 million reads were generated for 240 samples across three experiments involving seven vaginally-relevant bacteria. Taxonomic classification was performed using the STIRRUPS method and reference database [38]. Only 2,279 (<0.06%) below-threshold reads and 733 (<0.02%) above-threshold reads were assigned to taxa not in the study. None of the samples had large proportions of reads assigned to taxa not in the study (third quartile 0.02%, max 2.8%).
The three experiments consisted of mixing live bacteria, extracted DNA, and PCR product according to prescribed proportions (Additional file 2). Of the 80 samples in each experiment, 15 were technical replicates; i.e., there were 65 unique mixtures, 15 of which were repeated. The median absolute error, a measure of the technical variation among replicates, of the observed proportion of each organism was largest for the experiment mixing prescribed quantities of PCR product; the median absolute error was smallest for mixing live bacteria (Additional file 3). For all bacteria and experiments, the median absolute error was less than 5%. Therefore, technical variation was not a confounding factor in our results.
Our DNA extraction and PCR amplification protocols contributed more to bias than sequencing error and taxonomic misclassification
The results when mixing prescribed proportions of live bacteria, extracted DNA, and PCR product can be used to isolate the effects of the DNA extraction protocol, the PCR amplification protocol, and sequencing error and taxonomic classification.
Bacteria were collected from late log cultures and correlated with OD 600 nm to minimize the number of non-viable bacteria within the cultures. However, there was likely some contribution of DNA from non-viable bacteria and this would be expected to contribute to the bias. In these experiments, bias due to differing numbers of non-viable bacteria would not be distinguishable from bias due to our DNA extraction protocol. Throughout the remainder of the paper, it is implied that the bias due to DNA extraction includes bias due to non-viable bacteria.
A box plot of the bias for all mixtures for each of the three experiments and for each of the seven bacteria is plotted in Figure 2. The bias due to sequencing and classification was smallest, as indicated by the fact that the median bias was between -5% and 5% for each bacterium, and by the small inter-quartile ranges. The bias due to our DNA extraction protocol and our PCR amplification protocol contributed the most to total bias. Our observation that our PCR amplification protocol contributed more to bias than sequencing-specific causes confirms the results of a previous study [8]. Sequencing and classification error effects were likely reduced in our experiment because of the small numbers of bacteria in samples. Also, the use of a carefully-curated database and species-level classification method [38] likely helped to filter chimeric sequences.

Observed bias by bacterium. The observed bias (the observed minus the actual proportions) for each bacterium in the experimental design due to the different effects of our DNA Extraction, PCR amplification, and sequencing and taxonomic classification protocols. The total bias is also plotted for each bacterium. For each box and whisker plot, only the samples including the bacterium were included.
The effects of our DNA extraction and PCR amplification protocols were dependent on bacteria
The inter-quartile ranges for bias from our DNA extraction protocol indicated that our protocol amplified the observed proportions of L. crispatus, L. iners, P. bivia, and S. amnii while suppressing those of A. vaginae, S. agalactiae, and G. vaginalis. The same analysis for our PCR amplification protocol shows that the observed proportions of A. vaginae, L. iners, and S. amnii were increased while those of S. agalactiae, G. vaginalis, L. crispatus, and P. bivia were decreased. With the exception of bias due to our PCR amplification protocol and sequencing and classification for A. vaginae and P. bivia, the bias was significantly different from zero (p<0.05).
The effect of each processing step (DNA extraction, PCR amplification, sequencing and taxonomic classification) was dependent on the bacterium. The bias due to the different processing steps appeared to be independent because the bias from each step was cumulative and was reflected in the results for the total bias for each bacterium. The total bias is the observed proportion minus the proportion of cells included in the mixture in the first experiment. Overall, the observed proportions of A. vaginae, S. agalactiae, G. vaginalis, and L. crispatus was less than the proportions of bacteria in the mixtures, and the observed proportions of L. iners and S. amnii was more than the proportions in the mixtures. The inter-quartile range for total bias for mixtures containing P. bivia was large and contains zero, indicating that the bias was affected by which particular bacteria were also included in a mixture. The median total bias for S. agalactiae was -32.6%, indicating that in a mixture containing S. agalactiae, we would expect to observe that its proportion would be 32.6% less than its true representation in the community. The largest positive median bias among the bacteria was 39.1% for S. amnii.
Previous studies observed that the copy number and genome size may not be consequential sources of bias [19,33]. The analysis here appears to agree that these factors were not sufficient to describe bias in the PCR step. For example, S. agalactiae had the largest copy number among the organisms in this experiment, but the observed proportions were consistently less than the actual proportions in the mock communities.
Observed proportions of bacteria were amplified or suppressed by the presence of other bacteria
The interquartile range for bias due to our DNA extraction protocol was larger than that for bias due to our PCR amplification protocol or sequencing and classification for all but one bacterium (Figure 2), but the technical variation was largest when mixing pure PCR product (Additional file 3). Therefore, the wider ranges of bias due to our DNA extraction protocol was likely due to some relationship between bacterial signals. Here we present a deeper analysis of the effects observed at each step.
In traditional mixture experiment terminology, an interaction is a causative effect in the observed proportions of bacteria that may or may not reflect a physical relationship between the bacteria. The effect could be a result of a difference in proclivity to use resources. For example, one bacterium could yield more sequences in PCR product than another because of template re-annealing or primer design. For a bacterium A, there is a synergistic interaction with bacterium B if the presence of bacterium B increases the observed proportion of bacterium A. Likewise, there is an antagonistic interaction with bacterium B if the presence of bacterium B decreases the observed proportion of bacterium A. Because the term “interaction” is often interpreted in common use to connote a physical effect, we attempt to avoid the confusion and use the terms “relationship”, “synergistic relationship”, and “antagonistic relationship”.
Comparison of the prescribed proportions of bacteria with the results of the experiment mixing live bacteria can be used to evaluate whether the observed proportions of bacteria are promoted or suppressed by the presence of other bacteria. Special cubic mixture effect models, where the dependent variable is the observed proportion of a bacterium, reveal statistically significant blends of bacteria (Additional file 4). Each model has linear terms that capture the main effects of having bacteria present in the sample along with quadratic and cubic blending terms that capture higher-order relationships.
The model fits were clearly strong as indicated by R 2 values above 0.99 for each model. As expected from the results in Figure 2, there were far fewer statistically significant blending terms for the models based on mixing equal amounts of PCR product than for those based on mixing equal numbers of cells or those based on mixing equal quantities of DNA (Table 1, Additional file 4). The binary blends tended to be more statistically significant than ternary blends. The number of significant blending terms decreased for each subsequent experiment, as would be expected because the sources of bias due to our DNA extraction and PCR amplification protocols were removed. The interaction terms that were significant for the models for mixing cells (Experiment 1) but not significant for the models for mixing DNA (Experiment 2) highlight relationships for our DNA extraction protocol. Similarly, those terms that were significant for the models for mixing DNA (Experiment 2) but not significant for the models mixing PCR product (Experiment 3) highlight relationships for our PCR amplification protocol.
The models for the experiment mixing cells generated interaction/blending plots as shown in Figure 3. As an example, Figure 3(a) depicts the blending of L. crispatus and G. vaginalis. For a given concentration of L. crispatus, as more G. vaginalis is added to a sample (keeping absolute input quantities from other bacteria constant), the expected proportion of observed L. crispatus will increase. Therefore, G. vaginalis interacts synergistically with L. crispatus. The hypoteneuse of the triangle reflects the expected results when L. crispatus and G. vaginalis are the only bacteria in a mixture. The shaded contours if the remainder of the triangle indicate the expected observed L. crispatus proportion, averaging over the effects of including the other bacteria in the experiment at proportions resulting in a mixture that sums to 100%. The figure, therefore, depicts the expected relationship between two bacteria when included in blends at different levels.

Interaction/blending plots for L. crispatus and (a) G. vaginalis and (b) S. amnii . The contours indicate the expected observed amount of L. crispatus for a given actual percentage of a sample for a pair of bacteria.
Figure 3(b) depicts the antagonistic blending of S. amnii with L. crispatus: as more S. amnii are added to a sample with L. crispatus, the expected proportion of observed L. crispatus will decrease. These plots reflect properties that were quantified for statistical significance in the models (Additional file 3). For the model based on mixing equal numbers of cells, for which the observed proportion of L. crispatus is the dependent variable, the binary blend with G. vaginalis was significant with a coefficient estimate of 1.41, indicating that if L. crispatus and G. vaginalis were present in a sample together, then the proportion of observed L. crispatus would larger than what is truly present. This term in the model contributes 1.41 times the product of the actual proportions of L. crispatus and G. vaginalis to the expected amount of L. crispatus observed. For the same model, the binary blend for L. crispatus and S. amnii was significant with a negative coefficient estimate (-1.66), indicating that the observed proportion of L. crispatus decreases with increases in the proportion of S. amnii. If we compare this model with the model for mixing DNA, we see that the binary blend with G. vaginalis was not significant, but the binary blend with S. amnii was significant. These results indicated that during our DNA extraction process, the observed amount of L. crispatus will be increased relative to G. vaginalis and decreased relative to S. amnii. During our PCR amplification process, there was additional bias decreasing the observed amount of L. crispatus relative to S. amnii, but no significant relationship with G. vaginalis existed at this step.
Additional file 5 depicts the ratio of the observed L. crispatus to actual L. crispatus versus the proportion of G. vaginalis and S. amnii. The trend lines and the departures from y=1.0 help to indicate the blending effect. The wide variation in the data around the model indicated that the identities of the additional bacteria in a mixture were important for predicting the observed proportions of L. crispatus. Tables 2 and 3 contain the statistically significant synergistic and antagonistic binary blends for the experiment based on mixing equal numbers of cells. The significant relationships are ordered by decreasing significance. Nearly all of the bacteria were synergistic with L. iners and S. amnii, and nearly all are antagonistic with G. vaginalis. G. vaginalis was synergistic with all bacteria except S. agalactiae. All bacteria were antagonistic with S. agalactiae. These results indicate that the observed proportions of L. iners and S. amnii in samples were overestimates while those for G. vaginalis and S. agalactiae were underestimates.
Figure 4 presents the results of binary blends of equal amounts of cells, DNA, and PCR product for L. crispatus and S. agalactiae. When equal numbers of cells were mixed, over 92.5% of reads were assigned to L. crispatus, which is an error of 85%. When equal amounts of DNA were mixed, only 32% were assigned to L. crispatus. The discrepancy indicates that our DNA extraction protocol tends to increase the proportion of L. crispatus reads observed over that of S. agalactiae. The opposite trend was observed when comparing the results of mixing equal amounts of DNA and equal amounts of PCR product: the signal for S. agalactiae was increased and the signal for L. crispatus decreased during PCR. When equal amounts of PCR product were mixed, the observed proportions of the two bacteria were nearly equal, indicating that sequencing and taxonomic misclassification error did not contribute to observed bias.

Results for mixture of L. crispatus and S. agalactiae . Actual and observed proportions of bacteria when mixing equal proportions of cells (Exp. 1), DNA (Exp. 2), and PCR product (Exp. 3) for L. crispatus and S. agalactiae.
Among most samples with equal amounts of cells, rank abundance in the biased results was preserved. In other words, if for a pair of bacteria present in a sample the first is observed to comprise a larger proportion than the second, then the observed proportion for the first bacterium is larger than the second in most of the other samples containing those bacteria. A notable exception is depicted in Table 4. Sample 1 contained equal proportions of S. amnii, L. iners, and S. agalactiae. Because of bias, more S. amnii than L. iners was observed in Sample 1 in Table 4, and very little S. agalactiae was detected. Sample 2 also contained equal proportions of S. amnii and L. iners along with G. vaginalis and P. bivia. For this sample, more L. iners was observed than S. amnii. The differences were larger than the technical variation depicted in Additional file 2. Therefore, rank abundance was not preserved for L. iners and S. amnii. It is not clear whether the result was due to the larger number of bacteria in the samples than other samples, which is more representative of clinical and environmental samples, or was a reflection of tertiary and higher-order effects between combinations of bacteria.
Mixture effect models predicted community composition in clinical samples
Models constructed via an inverse fit (i.e., treating the mixing proportions of bacteria in a sample as the dependent variable) can be applied to the observed proportions of bacteria in clinical samples to estimate the true community composition. Figure 5 depicts the observed and predicted proportions of bacteria for samples from the mid-vaginal wall of a subject over the course of four visits to a clinic. Low diversity and richness in vaginal samples, such as that observed for this subject, is not unusual [39].

(a) Observed and (b) predicted proportions of bacteria of four clinical samples. The samples are from the same subject in different visits.
Because of the low diversity, vaginal samples are often classified by the predominant bacterium into community states or types [40]. The first sample, unadjusted, would be categorized as being of the L. iners type. However, the inverse models predicted that the true composition of the bacterial community was dominated by G. vaginalis. In our analysis of samples of the L. iners type from other subjects, we often observed subgroups consisting of those with nontrivial levels of G. vaginalis as in sample 1 and those with little as in sample 3 (unpublished results).
The bacteria common to the clinical samples and the mixture effect experiments were G. vaginalis, L. iners, and A. vaginae. G. vaginalis is often associated with a diagnosis of bacterial vaginosis (BV) and an elevated pH. L. iners and A. vaginae are lactic acid-producing bacteria known to lower pH. The observed proportion of G. vaginalis was larger for sample 2 than sample 4 and the proportion of lactic acid-producing bacteria was smaller, yet the pH was higher for sample 4. The predicted proportions aligned better with the pH measurements. Sample 3 was predicted to have the highest proportion of lactic acid-producing bacteria and was associated with the lowest pH, samples 1 and 2 had similar proportions of G. vaginalis and were associated with the same pH, and sample 4 had the largest proportion of G. vaginalis and was associated with the highest pH. Though samples 2 and 4 had the highest proportions of G. vaginalis and the highest measured pHs, there was no diagnosis. The lack of symptoms and/or disease may have been due to the presence of A. vaginae.
Conclusions
We have demonstrated that models based on analysis of small mock communities can enhance our understanding of bias and the analysis of low-diversity environments such as the human vagina. The same protocol could be applied to bacteria from more diverse environments such as the human gut or soil samples to understand how the most dominant species are affected by bias. As demonstrated here, a good understanding of bias can change and improve the conclusions based on the analysis of clinical or environmental samples.
We recommend that labs use small mock communities for understanding the effects of bias for their particular choices of protocols. Fitting mixture effect models is useful for establishing statistical significance concerning the relationship between observed proportions for pairs of bacteria. Though our results from a full mixture experiment and mixture effect models were informative, it is perhaps excessive for validating a pipeline. Smaller sets of mock communities, with around 10-20 blends of bacteria, are likely sufficient for labs to assess the effects of bias for certain taxa so that analysis may be qualitatively hedged. For example, if our lab observes clinical samples with both L. iners and G. vaginalis, we may presume that L. iners is likely over-estimated and G. vaginalis is under-estimated.
Additional experiments with axial blends and blends of larger numbers of bacteria are needed to understand if rank abundance is preserved. Using the protocol presented here to model bias for more diverse environments would require the creation and analysis of an impracticable number of mock communities because of the number of bacteria that need to be modeled. Therefore, it is necessary to further understand the factors contributing to bias from DNA extraction and PCR amplification protocols such as cell lysability, primer efficiency, Gram negativity, and GC content. Models based on these factors, along with careful curation of reference databases containing such information, could enhance the interpretation of results from studies of diverse communities.
The results presented here indicate that bias due to our DNA extraction and PCR amplification protocols are much greater than the effects of sequencing and taxonomic classification. Therefore, we can expect that bias will remain a challenge even as sequencing technology advances. The effects of bias can lead to the discovery of spurious correlations (linear relationships) and to missed true correlations. The results of ground-truthing with small mock communities can help to hedge conclusions obtained by analyzing observed relative quantities. Efforts to assess bias within labs as proposed here and guidelines for best practices across labs, a goal of The Microbiome Quality Control project [3], will facilitate the extraction of more useful information from experiments in various domains.
Methods
Mock community preparation and processing
The seven strains used, 16S rRNA gene copy numbers, and genome sizes are in Table 5. Copy numbers and genome sizes were estimated from NCBI [41]. Atopobium vaginae, Gardnerella vaginalis, Lactobacillus iners, Prevotella bivia, Streptococcus agalactiae, and Sneathia amnii were cultivated on Brain Heart Infusion (BHI) agar plates (EMD, Gibbstown, NJ) supplemented with 1% yeast extract, 2% gelatin, 0.1% starch, 0.1% glucose, and 10% human blood (sBHI) or in sBHI broth containing 10% human blood instead of serum [42]. Lactobacillus crispatus was grown on De Man Rogosa Sharpe (MRS) agar plates or in MRS broth. All bacteria were cultured at 37°C under anaerobic conditions (AnaeroPack, Mitsubishi Gas Chemical Co, Tokyo, Japan) until they reached late log phase. The optical density at 600 nm was determined and the bacteria were enumerated by counting colonies on solid medium. OD 600 nm values and colony forming units (CFUs) were determined in three separate experiments to ensure that the correlation between OD 600 nm values and CFU was precise for each species. Bacteria were aliquoted and kept frozen at -80°C until use. Bacteria were combined at the prescribed proportions based on CFU. DNA was extracted from the combinations using the PowerSoil DNA Isolation Kit from MO BIO Laboratories, Inc. (Carlsbad, California) and 2 μL of DNA was used in each PCR reaction.
For the DNA combinations, DNA was isolated from approximately 1×1010 bacteria using the Qiagen Genomic Tip 100/G kit (Valencia, CA) and DNA concentration and purity was measured using a Nanodrop (Thermo Fisher). The Qiagen kit was used only for extractions from pure cultures; using a different kit for these extractions would not create bias because the DNA are mixed at prescribed proportions after the extractions. The DNA from different species was combined to produce a final DNA concentration of 2 ng/ μL, and 2 μL of these dilutions were used in each PCR reaction.
For the PCR combinations, 4 ng of DNA from each organism was amplified by PCR and the PCR reactions were combined based on volume, according to the prescribed combinations.
The protocols of the Vaginal Human Microbiome Project for 16S rRNA gene sequencing have been previously described [38,43], and were followed for this study. The V1-V3 hypervariable regions of the bacterial 16S rRNA gene were amplified by PCR using barcoded primers. For each reaction, 4 ng of DNA was combined with 33 μL PCR Supermix High Fidelity™, 11 μL Platinum PCR Supermix™ (Life Technologies), and 100 nM each of forward and reverse primers. The 16S primers contain the A or B Titanium sequencing adapter (shown in italics), followed immediately by a unique variable (6-9 base) barcode sequence and finally the 20 nucleotide sequence complementary to the targeted region of the 16S rRNA gene. The forward primer was a mixture (4:1) of the primers Fwd-P1 (5’ - CCATCTCATCCCTGCGTGTCTCCGACTCAG BBBBBB AGAGTTYGATYMTGGCTYAG) and Fwd-P2 (5’ - CCATCTCATCCCTGCGTGTCTCCGACTCAG BBBBBB AGARTTTGATCYTGGTTCAG). The reverse primer was Rev1B (5’ – CCTATCCCCTGTGTGCCTTGGCAGTCTCAG ATTACCGCGGCTGCTGG). PCR products were sequenced using the Roche 454 GS FLX Titanium platform. The forward PCR primer is a mix of 20 different primers corresponding to positions 8 to 27 of the E. coli 16S rRNA genes. This primer mix contains primers that perfectly match the taxa used in this study with the exception of G. vaginalis with a single mismatch at position 19 (A/G) from the 3’-end of the forward primer.
Observed counts for the experiments mixing equal proportions of live bacteria were adjusted by dividing by 16S rRNA gene copy number. Observed counts for the experiments mixing equal proportions of DNA were adjusted by multiplying by genome size and dividing by copy number. Counts in each experiment were normalized to proportions.
The raw sequence data are available at http://www.ncbi.nlm.nih.gov/bioproject/267701 under BioProject ID PRJNA267701.
Species-level taxonomic classification
For both mock community samples and clinical samples, we processed reads for which valid primer and multiplex identifier sequences were observed, less than 10% of base calls had a quality score less than 10, the average quality score was greater than Q20, and the read length was between 200 and 540 bases. The reads were not trimmed. The STIRRUPS method for species-level taxonomic classification was used as previously described [38]. Sequences that aligned with at least 97% global sequence identity to a sequence in the reference database were classified. Across the three experiments, 253,078 reads (6.5%) were below threshold and were not included in subsequent analysis. Of the below-threshold reads, 252,796 were assigned to taxa in the study.
Experimental design and mixture effect models
With a mixture experiment, the levels of the individual bacteria strains cannot be set independently because the community proportions must sum to one. Suppose there are p bacteria and x i denotes the proportion of the i th bacterium in a mock community. Then, the x i s are constrained such that x i ≥0 for i=1,2,…,p and that
Due to the constraint that each treatment combination must sum to one, the form of mixture effect polynomials is somewhat different from the standard polynomials used in response surface modeling. In particular, we made use of Scheffé mixture effect models [36]. It is often the case in mixture experiments that a higher-order response surface model is needed to adequately model the response. We used a special cubic model of the form
Each coefficient β i represents the expected response when x i =1 and all other components are zero (i.e., pure blends). The β ij and β ijk are binary and ternary blending coefficients. For instance, each β ij is a measure of the departure from linearity when bacteria i and j are blended together. The sign of the nonlinear blending coefficients indicate whether the relationship is synergistic (positive coefficient) or antagonistic (negative coefficient). Models were fit using JMP [46]. Additional file 6 contains a JMP scripting language (JSL) script for fitting mixture effect models.
Quantifying contribution to bias
The differences in Figure 2 between observed and actual proportions were included only for mixtures in which the bacteria were present.
To test for statistical significance of bias, samples were matched between pairs of experiments (Experiments 1 and 2, 2 and 3, and 3 and the prescribed mixing ratios). Bootstrap confidence intervals for the Mahalanobis distance were calculated for each pair, and significance was established if zero was in the confidence interval. The Mahalanobis distance is a measure that accounts for correlations in the data so that distances along directions of low variation are larger and vice versa. Figures were generated and tests conducted using the R Environment for Statistical Computing and the packages bootstrap [44] and ggplot2 [45]. R code and a guide to data and scripts is contained in Additional file 7. Additional data and scripts are contained in Additional files 8, 9, 10, 11, 12, 13, 14 and 15.
Clinical samples and inverse models
Clinical samples were collected as part of the Vaginal Human Microbiome Project (VaHMP). Protocols for subject enrollment and sample collection have been previously described [38,43].
Inverse models were constructed to predict actual proportions based on observed proportions of bacteria in samples where equal quantities of cells were mixed. The dependent variable in these models is a scaling factor: the observed proportion divided by the actual proportion (observations are created for nonzero actual proportions only). The predictors were the actual proportions in each sample. One model was constructed for each bacterium. The models were used to make predictions of scaling factors for bacteria in clinical samples with non-zero counts. The observed values were adjusted for copy number and then multiplied by the predicted scaling factors to yield predictions for the actual proportions. These predictions were re-normalized to sum to 100%. Each model was a random forest model built with 500 trees and two splitting variables in each tree using the R package randomForest [47]. R code is contained in Additional file 7.
Processing of jumpstart consortium mock community
The composition and analysis of the Human Microbiome Project Jumpstart Consortium Mock Community have been previously described [18,31]. DNA Extraction, PCR amplification, and taxonomic sequencing were conducted as described for the other experiments. Taxonomic classification was performed using the Ribosomal Data Project Classifier [32] with a confidence threshold of 80%.
References
Lagier J-C, Million M, Hugon P, Armougom F, Raoult D. Human gut microbiota: Repertoire and variations. Front Cell Infect Microbiol. 2012; 2:136.
Knight R, Jansson J, Field D, Fierer N, Desai N, Fuhrman JA, et al. Unlocking the potential of metagenomics through replicated experimental design. Nat Biotech. 2012; 30(6):513–20.
The Microbiome Quality Control Project (MBQC). [http://www.mbqc.org]
Pinto AJ, Raskin L. PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS ONE. 2012; 7:43093.
Hong SH, Bunge J, Leslin C, Jeon S, Epstein SS. Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J. 2009; 3:1365–73.
Ahn J-H, Kim B-Y, Song J, Weon H-Y. Effects of PCR cycle number and DNA polymerase type on the 16S rRNA gene pyrosequencing analysis of bacterial communities. J Microbiol. 2012; 50:1071–4.
Lagier J-C, Armougom F, Million M, Hugon P, Pagnier I, Robert C, et al. Microbial culturomics: Paradigm shift in the human gut microbiome study. Clin Microbiol Infect. 2012; 18:1185–93.
Lee CK, Herbold CW, Polson SW, Wommack KE, Williamson SJ, McDonald IR, et al. Groundtruthing next-gen sequencing for microbial ecology-biases and errors in community structure estimates from PCR amplicon pyrosequencing. PLoS ONE. 2012; 7:44224.
Wu J-Y, Jiang X-T, Jiang Y-X, Lu S-Y, Zou F, Zhou H-W. Effects of polymerase, template dilution and cycle number on PCR based 16S rRNA diversity analysis using the deep sequencing method. BMC Microbiology. 2010; 10:255.
Wu G, Lewis J, Hoffmann C, Chen Y-Y, Knight R, Bittinger K, et al. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiology. 2010; 10(1):206. M3: 10.1186/1471-2180-10-206.
Kanagawa T. Bias and artifacts in multitemplate polymerase chain reactions (PCR). J Biosci Bioeng. 2003; 96:317–23.
Feinstein LM, Sui WJ, Blackwood CB. Assessment of bias associated with incomplete extraction of microbial DNA from soil. Appl Environ Microbiol. 2009; 75:5428–33.
Whitehouse CA, Hottel HE. Comparison of five commercial DNA extraction kits for the recovery of Francisella tularensis DNA from spiked soil samples. Mol Cellular Probes. 2007; 21:92–6.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009; 75:7537–41.
Quince C, Lanzén A, Curtis TP, Davenport RJ, Hall N, Head IM, et al. Accurate determination of microbial diversity from 454 pyrosequencing data. Nature Methods. 2009; 6:639–641.
Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. Wrinkles in the rare biosphere: Pyrosequencing errors can lead to artificial inflation of diversity estimates. Environmental Microbiology. 2010; 12:118–123.
Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environmental Microbiology. 2010; 12:1889–1898.
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011; 21:494–504.
Kembel SW, Wu M, Eisen JA, Green JL. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Computational Biology. 2012; 8:1002743.
Dubourg G, Lagier J-C, Armougom F, Robert C, Hamad I, Brouqui P, et al. The gut microbiota of a patient with resistant tuberculosis is more comprehensively studied by culturomics than by metagenomics. Eur. J. Clin. Microbiol. Infect. Dis. 2013; 32:637–645.
Paulson JN, Stine OS, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nature Methods. 2013; 10:1200–1202.
Bergmann GT, Bates ST, Eilers KG, Lauber CL, Caporaso JG, Walters WA, et al. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biology & Biochemistry. 2011; 43:1450–1455.
Lauber CL, Hamady M, Knight R, Fierer N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Applied and Environmental Microbiology. 2009; 75:5111–5120.
Andreson R, Mols T, Remm M. Predicting failure rate of PCR in large genomes. Nucleic Acids Res. 2008; 36:66.
Shinoda N, Yoshida T, Kusama T, Takagi M, Hayakawa T, Onodera T, et al. High GC contents of primer 5’-end increases reaction efficiency in polymerase chain reaction. Nucleosides, Nucleotides, and Nucleic Acids. 2009; 28:324–330.
Kiviharju K, Leisola M, Eerikäinen T. Optimization of Streptomyces peucetius var. caesius n47 cultivation and ε-rhodomycinone production using experimental designs and response surface methods. Journal of Industrial Microbiology and Biotechnology. 2004; 31:475–481.
Rispoli FJ, Shah V. Mixture design as a first step for optimization of fermentation medium for cutinase production from Ceolletotrichum lindemutianum. Journal of Industrial Microbiology and Biotechnology. 2007; 5:349–355.
Bautista-Gallego J, Arroyo-López FN, Chiesa A, Duráin-Quintana MC, Garrido-Fernández A. Use of a D-optimal design with constrains to quantify the effects of the mixture of sodium, potassium, calcium and magnesium chloride salts on the growth parameters of Saccharomyces cerevisiae. Journal of Industrial Microbiology and Biotechnology. 2008; 35:889–900.
Harbi B, Chaieb K, Jabeur C, Mahdouani K, Bakhrouf A. PCR detection of nitrite reductase genes (nirk and nirs) and use of active consortia of constructed ternary adherent staphylococcal cultures via mixture design for a denitrification process. World Journal of Microbiology and Biotechnology. 2010; 31:473–480.
Arroyo-López FN, Bautista-Gallego J, Chiesa A, Durán-Quintana MC, Garrido-Fernández A. Use of a D-optimal mixture design to estimate the effects of diverse chloride salts on the growth parameters of Lactobacillus pentosus. Food Microbiology. 2009; 26:396–403.
Jumpstart Consortium Human Microbiome Project Data Generation, Working Group. Evaluation of 16S rDNA-based community profiling for human microbiome research. PLoS ONE. 2012; 7(6):39315. M3: doi:10.1371/journal.pone.0039315.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology. 2007; 73(16):5261–5267.
Polz MF, Cavanaugh CM. Bias in template-to-product ratios in multitemplate PCR. Applied and Environmental Microbiolology. 1998; 64:3724–3730.
Huber JA, Morrison HG, Huse SM, Neal PR, Sogin ML, Welch DBM. Effect of PCR amplicon size on assessments of clone library microbial diversity and community structure. Environmental microbiology. 2009; 11(5):1292–1302.
Cornell J. Experiments with Mixtures: Designs, Models, and the Analysis of Mixture Data. New York: Wiley; 2002.
Scheffé H. Experiments with mixtures. Journal of the Royal Statistical Society, Series B. 1958; 20:344–366.
Goos P, Jones B. Optimal Design of Experiments: A Case Study Approach. New York: Wiley; 2011.
Fettweis JM, Serrano MG, Sheth NU, Mayer CM, Glascock AL, Brooks JP, et al. Species-level classification of the vaginal microbiome. BMC Genomics. 2012; 13:17.
The Human Microbiome Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012; 486:207–214.
Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proceedings of the National Academy of Sciences. 2011; 108(Supplement 1):4680–4687.
National Center for Biotechnology Information (NCBI). [http://www.ncbi.nlm.nih.gov]
Harwich MD Jr., Serrano MG, Fettweis JM, Alves JM, Reimers MA, Vaginal Microbiome Consortium (additional members), et al. Genomic sequence analysis and characterization of Sneathia amnii sp. nov. BMC Genomics. 2012; 13 Suppl 8:4–21641384201217.
Fettweis JM, Alves JP, Borzelleca JF, Brooks JP, Friedline CJ, Gao Y, et al. The vaginal microbiome: Disease, genetics and the environment. Nature Precedings, 10–1038201151502 (2011).
Leisch F. boostrap: Functions for the Book “An Introduction to the Bootstrap”. [http://cran.rproject.org/web/packages/bootstrap/index.html]
Wickham H. Ggplot2: Elegant Graphics for Data Analysis. New York: Springer; 2009.
JMP. [http://www.jmp.com]
Liaw A, Wiener M. Classification and regression by randomforest. R News. 2002; 2:18–22.
Acknowledgements
This study was supported in part by NIH awards UH3AI08326-01, 2P60MD002256-06, 8U54HD080784; an award from the The Thomas F. and Kate Miller Jeffress Memorial Trust, Bank of America, Trustee; and NSF grant DMS-1127914 to the Statistical and Applied Mathematical Sciences Institute. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. Sequence analysis was performed in the Nucleic Acids Research Facilities at VCU. Bioinformatics analysis was provided by the staff of the Bioinformatics Computational Core Laboratories at VCU.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JPB conceived the experiment, analyzed data, and wrote the paper; DJE designed the experiment, analyzed data, and helped write the paper; MDH carried out the experiment and contributed specialized knowledge related to growing bacteria; MCR, JMF, KKJ, and GAB helped design the experiment; MGS, NUS, and BH conducted bioinformatic analyses of sequence data; RAR analyzed data; PG, JFS, and KKJ provided analyses of results from clinical samples; all authors contributed conceptual advice in conducting the experiment and writing the paper. All authors read and approved the final manuscript.
Authors’ information
Vaginal Microbiome Consortium (additional members). Vaginal: vmc.vcu.edu, Virginia Commonwealth University, 23284, Richmond, VA, USA.
Additional files
Additional file 1
Comparison of DNA extraction kits and number of PCR cycles. Stacked bar plot of observed proportions of bacteria for a mock community consisting of 21 strains of bacteria when using PowerSoil and Qiagen DNA extraction kits and when allowing 30 and 35 PCR cycles.
Additional file 2
Experimental design. Table of the prescribed mixing proportions, plate, and barcode for the experiments mixing equal proportions of cells, DNA, and PCR product.
Additional file 3
Plot of technical variation in observed proportions of bacteria. Boxplot of the technical variation, measured as the absolute difference between observed proportions in replicate samples, for each bacterium and for experiments mixing equal proportions of cells, DNA, and PCR product.
Additional file 4
Tables of significant blending terms for mixture effect models. Coefficient estimates, t ratios, and p values for significant cofficients of mixture effect models for each bacterium and for experiments mixing equal proportions of cells, DNA, and PCR product.
Additional file 5
Alternate visualization of Figure 3 with data. (left) The ratio of observed to actual L. crispatus versus the actual G. vaginalis for the samples, and the expected values based on mixture effect models. (right) The ratio of observed to actual L. crispatus versus the actual S. amnii for the samples, and the expected values based on mixture effect models.
Additional file 6
JMP Scripting Language file. Script for fitting mixture effect models for the experiments mixing equal amounts of cells, DNA, and PCR product.
Additional file 8
Taxonomic classification of reads. Taxonomic classification of reads from experiments mixing equal amounts of cells, DNA, and PCR product. This file is the output of the STIRRUPS [38] pipeline.
Additional file 9
Python script for creating counts tables. Python script for creating tables of above-threshold and below-threshold counts for the reads from the experiments mixing equal amounts of cells, DNA, and PCR product.
Additional file 10
Table of above-threshold counts. Above-threshold counts for each sample in the experiments mixing equal amounts of cells, DNA, and PCR product.
Additional file 11
Table of below-threshold counts. Below-threshold counts for each sample in the experiments mixing equal amounts of cells, DNA, and PCR product.
Additional file 12
Design and results for experiment mixing cells. Prescribed mixing proportions and observed proportions for the experiment mixing equal numbers of cells.
Additional file 13
Design and results for experiment mixing DNA. Prescribed mixing proportions and observed proportions for the experiment mixing equal amounts of DNA.
Additional file 14
Design and results for experiment mixing PCR product. Prescribed mixing proportions and observed proportions for the experiment mixing equal amounts of PCR product.
Additional file 15
File containing information about the classification of each read for the experiments mixing equal amounts of cells, DNA, and PCR product. The columns are sample name, read name, RDP classification, RDP score, RDP classification level, STIRRUPS species classification, above threshold (AT) or below threshold (BT) STIRRUPS classification, STIRRUPS score.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Brooks, J.P., Edwards, D.J., Harwich, M.D. et al. The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol 15, 66 (2015). https://doi.org/10.1186/s12866-015-0351-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-015-0351-6