
Abstract
Background
Burkholderia pseudomallei is a facultative intracellular pathogen and the causative agent of melioidosis. A conserved type III secretion system (T3SS3) and type VI secretion system (T6SS1) are critical for intracellular survival and growth. The T3SS3 and T6SS1 genes are coordinately and hierarchically regulated by a TetR-type regulator, BspR. A central transcriptional regulator of the BspR regulatory cascade, BsaN, activates a subset of T3SS3 and T6SS1 loci.
Results
To elucidate the scope of the BsaN regulon, we used RNAseq analysis to compare the transcriptomes of wild-type B. pseudomallei KHW and a bsaN deletion mutant. The 60 genes positively-regulated by BsaN include those that we had previously identified in addition to a polyketide biosynthesis locus and genes involved in amino acid biosynthesis. BsaN was also found to repress the transcription of 51 genes including flagellar motility loci and those encoding components of the T3SS3 apparatus. Using a promoter-lacZ fusion assay in E. coli, we show that BsaN together with the chaperone BicA directly control the expression of the T3SS3 translocon, effector and associated regulatory genes that are organized into at least five operons (BPSS1516-BPSS1552). Using a mutagenesis approach, a consensus regulatory motif in the promoter regions of BsaN-regulated genes was shown to be essential for transcriptional activation.
Conclusions
BsaN/BicA functions as a central regulator of key virulence clusters in B. pseudomallei within a more extensive network of genetic regulation. We propose that BsaN/BicA controls a gene expression program that facilitates the adaption and intracellular survival of the pathogen within eukaryotic hosts.
Similar content being viewed by others

Background
Melioidosis is a serious and often fatal infectious disease common to Southeast Asia and Northern Australia caused by the Gram-negative soil bacterium Burkholderia pseudomallei. B. pseudomallei is a highly versatile pathogen capable of surviving inside mammalian cells and in many environmental niches. The bacterium can infect numerous animal species, amoebae, nematodes, and tomato plants [1]–[5], and has been previously found within the tissues of exotic grasses in Australia [6]. The environmental origin of B. pseudomallei and its promiscuous host range have shaped the hypothesis that some of its genetic loci evolved in the rhizosphere as anti-predation determinants that subsequently promote “accidental” virulence in humans and animals. In recent years, important advances have been made in understanding the pathogenic mechanisms of B. pseudomallei including the roles of the Type III and Type VI Secretion Systems (T3SS, T6SS) [7]–[11]. B. pseudomallei contains three T3SSs and six T6SSs, but only T3SS3 (also referred to as the Burkholderia secretion apparatus, or T3SSBsa) and T6SS1 are critical for pathogenesis in mice and hamsters [7],[12],[13].
Expression of the T3SS3 and T6SS1 gene clusters is tightly controlled, both temporally and spatially, during the B. pseudomallei intracellular lifecycle. We have identified a regulatory cascade that coordinately activates T3SS3 and T6SS1 gene expression in growth medium and in infected mammalian cells [8],[14]. At the top of the cascade is the TetR-type regulator BspR that stimulates the expression of bprP. The bspR gene is located on chromosome 1 of the B. pseudomallei genome whereas bprP is part of the T3SS3 gene cluster on chromosome 2 [14]. The ToxR-like BprP in turn activates genes encoding the structural components of T3SS3, including the araC-type regulatory gene bsaN. BsaN is important for the activation of T3SS3 effector and translocon gene expression, and several regulatory genes including bprC and virAG, whose gene products control T6SS1 expression [8]. The mechanisms through which these transcriptional regulators control the expression of their target genes are not understood. It is also unclear whether these regulators are acting directly on the identified target genes or through as yet undiscovered intermediary regulators, and whether additional host cell cofactors are involved that may serve as intracellular signals.
Compared to T3SSs in other pathogens such as Pseudomonas, Salmonella and Shigella, only a limited number of effectors have been identified for B. pseudomallei T3SS3. One of the effector proteins secreted by T3SS3 is BopE, which is annotated to exhibit guanine nucleotide exchange factor activity and has been reported to facilitate invasion of epithelial cells [15]. bopA is generally assumed to encode a T3SS3 effector since it is located adjacent to bopE, although T3SS3-dependent secretion of BopA has never been verified. Functionally, BopA has been described to promote resistance to LC3-associated autophagy and a bopA mutation results in an intracellular replication defect [16],[17]. A third effector protein, BopC (BPSS1516), was recently shown to be secreted via T3SS3, and bopC mutants were reported to be less invasive in epithelial cells [18] and to exhibit delayed endosome escape and reduced intracellular growth in J774 murine macrophages [19].
To determine the full extent of the BsaN regulon and examine whether BsaN activates the expression of additional effector genes, we performed global transcriptome analysis of B. pseudomallei KHW wildtype (WT) and a ΔbsaN mutant strain using RNAseq. Our analysis shows that 111 genes are under the direct or indirect transcriptional control of BsaN. In addition to activating loci associated with T3SS3, we demonstrate that BsaN functions to repress transcription of other loci. Thus, BsaN functions as a central regulatory factor within a more extensive network to facilitate the intracellular lifecycle of B. pseudomallei.
Results
Identification of the BsaN regulon through RNAseq analysis
BsaN (BPSS1546 in the reference B. pseudomallei K96243 genome) was previously shown to function as a central regulator of a hierarchical cascade that activates effector and translocon genes of T3SS3 as well as several associated regulatory genes [8],[14]. Furthermore, BsaN was shown to activate the expression of certain T6SS1-associated genes including the two-component regulatory system locus virAG (BPSS1494, 1495), and the bim actin motility genes (BPSS1490-1493). To gain further insight into the BsaN regulon, massive parallel sequencing was performed on cDNA prepared from RNA isolated from wild-type and ΔbsaN mutant strains. We had previously shown that complementation of our ΔbsaN mutant with a bsaN plasmid could restore the secretion of the BopE effector [14], showing that our complementation restored protein expression of the effectors and that the mutation was specific to bsaN and not due to off target effects.
Between 16 and 56 million reads (n = 2 from 3 combined cultures) were obtained that aligned to non-ribosomal genes in the KHW [20] genome (Additional file 1: Table S1). Reads of the technical replicates displayed high reproducibility (R-value) (Additional file 1: Table S1) demonstrating that variability was not introduced through sample preparation or sequencing errors. The K96243 reference genome was co-aligned for ease of gene annotation. The nucleotide sequences of chromosomes I and II are 99.3 and 99.1% identical, respectively.
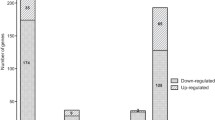
Comparison between wild-type and ΔbsaN transcriptomes identified 111 genes that were differentially regulated using 3-fold or more (adjusted p-value < 0.01) as the cut off. Of these, 60 genes were expressed more highly in wild-type KHW compared to the ΔbsaN strain, indicating that BsaN directly or indirectly activates their transcription (Table 1). However, 51 genes were expressed more highly in the ΔbsaN mutant suggesting that BsaN can function directly or indirectly as a repressor (Table 2). RNAseq results were validated using quantitative real time-PCR (qRT-PCR) analysis for select loci. RNAseq analysis identified all genes that we had previously shown to be activated by BsaN [8],[14] (Figure 1A and 1B, Table 1). The effector and chaperone genes bopE, bopA and bicP together with the regulatory gene bprD were amongst the highest activated genes (50-270-fold). In addition, two putative transposase genes separating the T3SS3 genes and the T6SS1 gene clusters were highly activated by BsaN (Table 1). Genes activated at lower levels (3-4-fold) include a hybrid non-ribosomal peptide synthase (NRPS)/polyketide synthase (PKS) locus consisting of 22 genes (BPSL0472-BPSL0493) unique to B. pseudomallei and B. mallei. NRPS/PKS systems are found in microbes and fungi, and are generally responsible for the production of complex natural compounds such as antibiotics and siderophores. Burkholderia species are rich in NRPS/PKS loci that contain multiple metabolic genes or encode large multidomain synthases [21]. Although the precise function of this NRPS/PKS locus is not currently known, the presence of a diaminobutyrate-2-oxoglutarate amino transferase gene (BPSL0476) suggests that 2,4-diaminobutrate is one of the polyketide’s component. Loci for methionine and threonine biosynthesis, as well as ribose uptake (Table 2), were activated at similar levels. Representative BsaN-activated genes were confirmed by qRT-PCR (Figure 1C-D).

Regulation of selected genes by BsaN as analyzed by RNAseq and qRT-PCR. A. Activation and repression of T3SS3 cluster genes as analyzed by RNAseq. The adjusted p value for all genes is less than 0.01 with the exception of three genes denoted with ^. B. Activation of BsaN regulated T6SS1 and bim motility genes as analyzed by RNAseq. C and D qRT-PCR validation of selected activated genes. Expression of each in wild-type B. pseudomallei KHW gene is set to 1; transcription was normalized to that of the recA reference gene. E. qRT-PCR validation of repressed genes. Expression of each in wild-type B. pseudomallei KHW gene is set to 1; transcription was normalized to that of the 16S rRNA reference gene. The flgL gene is located upstream and in the same transcriptional unit as flgK.
Intriguingly, genes encoding the T3SS3 apparatus components were found to be repressed in the wildtype compared with the ΔbsaN mutant, suggesting a role for BsaN in limiting apparatus synthesis when translocon and effector genes are transcribed (Figure 1A, 1E, Table 2). Also repressed are polar flagellar motility loci on chromosome 1 including the flagellin genes fliC and fliD, as well as flagellar hook proteins flgL and flgK. Repression of these genes as well as motA (BPSL3309) and cheD (BPSS3302) were validated by qRT-PCR (Figure 1E). In Salmonella and other bacteria, motAB are key components of the flagellar motor complex [22]. motAB in KHW are part of a chemotaxis (che) locus, which is repressed 2–2.9-fold (p < 0.01) as assessed by RNAseq. In addition, expression of a second polyketide biosynthesis locus (BPSS0303-BPSS0311) was reduced in a ΔbsaN mutant, possibly by repression of a co-localized araC-type regulatory gene, BPSS0336 (Table 2). However, down-regulation of this cluster could not be verified by qRT-PCR (data not shown). We were likewise unable to validate repression of BPSL2404-2405, which putatively encode transport and energy metabolism functions, respectively, in addition to BPSS1887-1888, which are postulated to encode oxidative enzymes for energy metabolism.
Additional loci implicated in lipid and energy metabolism are also repressed (Table 2). Catabolic genes encode a cytochrome o oxidase typically used by bacteria in an oxygen-rich environment [23], along with enzymes involved in the aerobic degradation of aromatic compounds and in the degradation of arginine. A gene involved in the synthesis of betaine, an osmoprotectant which serves to adapt Gram-negative bacteria to conditions of high osmolarity, is also repressed by BsaN.
BsaN together with chaperone BicA directly activate T3SS3 effector and T6SS1 regulatory genes
We have previously shown that expression of the two component regulatory system virAG and the genes from BPSS1520 (bprC) to BPSS1533 (bicA) in the T3SS3 cluster were regulated by BsaN in concert with the chaperone BicA [14]. To determine whether BsaN/BicA activate these genes directly, bsaN and bicA open reading frames (orfs) from B. pseudomallei strain KHW were inserted into a plasmid downstream of an arabinose-inducible promoter on pMLBAD [24]. These constructs were introduced into E. coli DH5α [25] along with an additional construct containing putative promoter regions of several BsaN target genes transcriptionally fused to lacZ on pRW50 [26] or pRW50mob, which contains the oriT fragment for pOT182 [27]. The effect of BsaN/BicA on promoter activity was then assessed by β-galactosidase activities.
The putative bsaN orf is annotated in the B. pseudomallei genome database to initiate from a GTG start codon [28]. We identified a second potential start codon (ATG) and ribosome binding site 117 nucleotides (nt) upstream of GTG (Figure 2A, B). bsaN/bicA expression constructs (Figure 2A) that were initiated from GTG were unable to activate transcription of bicA, bopA and bopE in E. coli (Additional file 1: Table S2), supporting the notion that the ATG was the actual start codon for BsaN. Furthermore, a transcriptional start site was identified 56 nucleotide upstream of the ATG codon via RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) (Figure 2B). A putative Ribosomal Binding Site (RBS) is located in front of the ATG condon. Replacing the GTG-initiated bsaN orf with the longer version containing the ATG start site resulted in activation of the bicA, bopA and bopE promoters as well as those for BPSS1521 (bprD), BPSS 1495 (virA) and the putative transposase BPSS1518 (Figure 3A-F). Expression of BsaN alone was not sufficient to activate these promoters (Additional file 1: Table S2), demonstrating the co-requirement for BicA. No apparent BsaN/BicA-dependent promoter activity was obtained for BPSS1528 (bapA), BPSS1523 (bicP), BPSS1530 (bprA), or BPSS1520 (bprC) (Additional file 1: Table S2) (refer to Figure 2C for gene location). Furthermore, BsaN/BicA could not activate transcription of a BPSS1512 (tssM)-lacZ fusion in E. coli (Figure 3G). Thus, BsaN/BicA drives the expression of bprDC and the BPSS1518-1516 operons directly, whereas bicP and bprB gene expression is likely driven by the upstream-located bopA promoter. Transcription of the bapABC and bprA genes could be driven from the bicA promoter. Collectively, these results are represented in Figure 2C where the five validated promoters and operon structures controlled directly by BsaN/BicA are depicted by black solid line arrows.

Transcriptional analysis of bsaN and BsaN/BicA-regulated genes. A. Schematic diagram of the bsaN gene. Arrow above +1 indicates the transcriptional start site and direction. Double-headed arrows indicated the DNA fragments used for the reconstitution of BsaN-mediated promoter activation experiments. B. Promoter region indicating the transcriptional start site and start codon of bsaN. Bold and underlined letter G indicates the transcriptional start site (+1 in 2A). Bold and underlined agga indicate the putative RBS. Bold and underlined ATG and GTG indicate the actual and wrongly annotated start codons of bsaN, respectively. C. Genetic and transcriptional organization of T3SS3 genes. Arrows indicate transcriptional units. Putative promoter regions are depicted as shaded spheres at the beginning of line arrows. Red line arrows denote operons regulated by BprP. Black line arrows indicate operons regulated by BsaN. Black dotted arrows with shaded diamonds represent putative promoters that were analyzed for direct activation by BsaN/BicA, however, no expression was found (Additional file 1: Table S2).

Activation of promoters by BsaN/BicA in E. coli. The ability of BsaN and BicA to directly activate the expression of promoters was examined by providing regulatory genes in trans and measuring β-galactosidase activities arising from the expression of transcriptional promoter-lacZ fusions in E. coli DH5α. Effect of BsaN/BicA on the expression of A. PbicA-lacZ fusion, B. PbopA-lacZ fusion, C. PbopE-lacZ fusion, D. PbprD-lacZ fusion and E. Ps1518-lacZ fusions; Ps1518 denotes the promoter region of BPSS1518. Effect of BsaN/BicA on the expression of F. PvirA-lacZ fusion and G. PtssM-lacZ fusion. *p < 0.05.
Identification of transcriptional start sites and the sequence motif for BsaN/BicA activation
Similarities between BsaN/BicA regulated promoters were examined by first determining their transcriptional start sites using RLM-RACE. One transcriptional start site was identified for the bicA, bprD and BPSS1518 promoters, and two start sites were detected for the bopA and virA promoters. We were unable to identify a transcriptional start site for bopE, which is divergently transcribed from bopA (Figure 2C). A 150-bp sequence upstream of each transcriptional start site was submitted to MEME (Motif Elicitation for Prediction of DNA Motifs), which identified a 15 bp motif that we designated as the putative BsaN box (Figure 4A). The distance from the transcriptional start site varied from 24 bp (virA) to 35 bp (bicA and bopA) (Figure 4B). When the motif was submitted to Motif Alignment & Search Tool (MAST) to search for other potential BsaN/BicA-regulated promoters in the B. pseudomallei genome (strain K96243), BsaN boxes were also found upstream of tssM and BPSS1889, a putative gene encoding an AraC family protein, in addition to those already identified. However, qRT- PCR analysis of BPSS1889 expression in ΔbsaN and ΔbicA mutants did not reveal a decrease in expression compared to wild-type bacteria (data not shown). BPSS1889 is located adjacent but transcribed in the opposite direction to the operon BPSS1884-1888, which was shown by RNAseq to be repressed by BsaN (Table 2). Although we could not confirm BsaN-dependent regulation of BPSS1889 by qRT-PCR, the upstream BsaN box suggests the possible involvement of this putative regulator in repression of the operon in vivo. It is likely that conditions for BsaN-dependent repression are difficult to establish in vitro resulting in variability and lack of validation. We also could not identify any −10 and −35 sequences for prokaryotic housekeeping sigma factor in these promoters. It is likely that the BsaN/BicA-regulated promoters are transcribed by one or more alternative sigma factors. Unfortunately, B. pseudomallei genome harbours more than 10 alternative sigma factors that have not been systematically studied. Therefore, their recognition sequences are currently unknown.

Sequence motifs in promoter regions of BsaN/BicA-regulated genes. A. The sequence motif for the BsaN box as indicated in bold, capital letters was identified using the bioinformatics tool MEME. B. The sequence of the BsaN box generated by MEME from the 5 BsaN-activating promoters as denoted in capital letters. The 3’capitalized letters denote the start of transcription with the exception of PtssM, which is the translational start codon of TssM.
tssM is one of the highly activated genes in our RNAseq analysis (Table 1) confirming previous in vivo expression studies [29]. However, despite the presence of the BsaN box upstream of the putative tssM operon (BPSS1512-1514), BsaN/BicA alone is not sufficient to activate tssM transcription in E. coli (Figure 3G). This suggests that tssM regulation is more complex and likely requires additional cis and/or trans-acting regulatory elements for activation.
Determining the sequence motif requirement for BsaN/BicA activation
To determine whether the putative BsaN box motif was required and sufficient for the other genes regulated by BsaN/BicA, we constructed two types of truncated promoter-lacZ fusions. The “type 1” deletion contained only the BsaN motif and lacked all upstream sequences. The “type 2” deletion lacked all upstream sequences in addition to the first six bp of the putative BsaN box motif. We assayed the ability of these truncated promoters to drive lacZ expression in the presence of BsaN/BicA. All truncated versions of the promoter regions for bicA, virA and BPSS1518 lost promoter activity (Figure 5A-C). In contrast, versions containing the intact BsaN box for bprD (Figure 5D) and bopA (Figure 5E) were still functional, but further truncation eliminated their activation. The type 1 truncated version of the bprD promoter (PbprD1-lacZ) had three-fold higher β-galactosidase activity than the full length promoter- lacZ fusion (PbprD-lacZ) (Figure 5D), whereas deletion of sequences upstream of the bopA promoter did not have a significant effect on the level of activation (Figure 5E).

Analysis of BsaN box requirements for transcription activation by BsaN/BicA. The ability of BsaN/BicA to directly activate the expression of truncated promoters was examined by providing regulatory genes in trans and measuring β-galactosidase activities arising from the expression of transcriptional promoter-lacZ fusions in E. coli DH5α. The top sequence of each gene includes the intact promoter region; sequence 1 is deleted up to the BsaN box; sequence 2 also includes a 6 nucleotide deletion of the BsaN box. Effect of BsaN/BicA on the expression of A. PbicA-lacZ fusion, B. PvirA-lacZ fusion and C. Ps1518-lacZ fusion; Ps1518 denotes the promoter region of BPSS1518. Effect of BsaN/BicA on the expression of D. PbprD-lacZ fusion and E. PbopA-lacZ fusion.
BprP directly activates bsaN and bsaM
In the hierarchical control of T3SS3 and T6SS1 expression, BspR was suggested to activate the expression of bprP[14]. Previously, BprP was shown to bind sequences upstream of bsaM and bsaN (refer to Figure 2C for gene location) [14], suggesting that it directly activates their transcription. bsaN is the first orf of the putative operon that encodes structural components of T3SS3 (Figure 2C) and is divergently transcribed from bsaM. To better understand how bsaN expression itself is controlled, we examined the relationships to its upstream regulators BspR and BprP using the LacZ fusion assay as described previously [8]. Plasmids with either bspR or bprP under arabinose induction control were introduced into E. coli containing plasmids with either a bsaN-lacZ fusion or a bsaM-lacZ fusion. A bprP-lacZ fusion served as control for BspR regulation. The ability of BspR and BprP to directly activate bsaN-lacZ, bsaM-lacZ and bprP-lacZ expression was determined by measuring β-galactosidase activity. As expected, BprP activated both the bsaM and bsaN promoters in E. coli (Figure 6A, B). The presence of bprQ, a gene immediate downstream from bprP, had no effect on the activity of BprP. Furthermore, BprP did not activate its own promoter in E. coli (data not shown). However, BspR was not able to activate the promoter of bprP demonstrating that this regulator is not active in E. coli or that additional factors are required for activation (Figure 6C).

Activation of bsaM and bsaN promoters by BprP in E. coli. The ability of BprP to directly activate the expression of promoters in the presence and absence of BprQ was examined by providing the bprP and bprQ genes in trans and measuring β-galactosidase activities arising from the expression of transcriptional promoter-lacZ fusions in E. coli DH5α. A. Effect of BprP and BprQ on the expression of PbsaN-lacZ fusion. B. Effect of BprP and BprQ on the expression of PbsaM-lacZ fusion. C. Effect of BspR on the expression of PbprP-lacZ fusion. *p < 0.05.
Analysis of BsaN/BicA-regulated virulence loci
BsaN/BicA directly induces the expression of three known T3SS3 effector loci, bopA, bopC and bopE. Recent studies suggest that the T3SS3 effectors BopC and BopE are involved in invasion of epithelial cells and endosome escape [15],[18],[19], while BopA has been implicated in escape from autophagy [17]. BopC was recently shown to be secreted via T3SS3 in B. pseudomallei K96243 [18], and our data confirm this (Additional file 1: Figure S1). In B. pseudomallei KHW, mutation of bopA, bopC or bopE[30] individually resulted in no detectable difference in numbers of bacteria inside RAW264.7 mouse macrophages when measured 2 hr. after infection (Additional file 1: Figure S2A). Upon extended incubation times, however, the ΔbopA and the ΔbopACE[30] strains exhibited an intracellular replication defect that was intermediate between levels observed for wildtype KHW and the ΔbsaM[30] or ΔbsaN mutant derivatives. No differences in intracellular growth or host cell cytotoxicity were observed for the bopC or bopE mutant strains, although infection with the bopA or bopACE triple deletion mutants resulted in a decrease in cytotoxicity (Additional file 1: Figure S2B) that coincided with a reduction in the rate of intracellular replication (Additional file 1: Figure S2A), suggesting that intracellular replication results in host cell toxicity. This is in contrast to the T3SS3 ΔbsaM and the ΔbsaN regulatory mutants in strain KHW, which are limited in their ability to multiply intracellularly as previously reported (Additional file 1: Figure S2A).
Three BsaN/BicA-activated orfs are located between the T3SS3 and T6SS1 loci, and upstream of the T3SS3 effector gene bopC. We analyzed these orfs for potential roles in intracellular replication and cell-to-cell spread. BPSS1512 encodes TssM, was previously shown to be secreted independently of T3SS3 and T6SS1 and functions as a broad-base deubiquitinase, with activity on TNFR-associated factor-3, TNFR-associated factor-6, and IκBα [31]. BPSS1513 is predicted to encode a short (97 aa) protein of unknown function and was not secreted under our assay conditions (Additional file 1: Figure S3A). folE (BPSS1514) encodes a putative GTP cyclohydrolase I, suggesting a role in tetrahydrofolate biosynthesis rather than in virulence. Consistent with this notion, Δ(BPSS1513-folE) mutant did not exhibit defects in cell-based virulence assays (Additional file 1: Figure S3B-E).
Discussion
T3SSs and T6SSs play important roles in bacterial-host cell interactions [32],[33]. As each system is a complex structure encoded by 20 or more genes, it is expected that their expression and assembly would be tightly regulated. In B. pseudomallei, T3SS3 and T6SS1 gene clusters are highly induced following host cell infection [8], and their function is critical for virulence in animal models [8],[13]. T3SS3 has been shown to promote escape from endocytic vesicles, and T6SS1 plays a key role in promoting intercellular spread by fusion of adjacent cell membranes, leading to the formation of MNGCs that can be found in melioidosis patients [34]. Upregulation of T3SS3 is mediated by a signalling cascade initiating from BspR through BprP, which in turn increases the expression of the AraC-type regulator BsaN. Using global transcriptome and promoter activation analysis, we have shown that the BsaN regulon occupies a central position in modulating the expression of T3SS3, T6SS1 and several additional loci that are likely involved in promoting virulence and intracellular survival.
Regulatory factors may act to control expression by acting directly on a given gene, or indirectly by modulating a regulatory intermediate. We found that BsaN in complex with the T3SS3 chaperone BicA directly controls the expression of 19 loci in a region of chromosome 2 containing T6SS1 and T3SS3 accessory genes (BPSS1494-BPSS1533). BsaN/BicA activated transcription of the operons encoding T3SS3 effector proteins, the BipBCD translocon complex, chaperones, and other transcriptional regulators, as well as two genes of unknown function (BPSS1513-1514). BsaN/BicA upregulates expression of T6SS1 by activating the transcription of the two component regulatory system loci virAG and bprC, which in turn induce the hcp and tssAB loci, encoding T6SS1 tube and sheath proteins [8],[35]. Interestingly, our RNAseq and qRT-PCR analyses revealed that BsaN also acts to repress transcription of T3SS3 apparatus genes in the bsaM and bsaN operons that are otherwise directly activated by the upstream regulator BprP. It is possible that BsaN mediates repression indirectly as the bsaM and bsaN intergenic region lacks a recognizable BsaN binding motif (see below). It is unlikely, however, that repression occurs due to decreased expression of bprP since its transcription is unchanged in a ΔbsaN mutant. Taken together, these findings demonstrate that BsaN plays a dual role in the regulation of T3SS3; one in coordinating translocon and effector transcription, and a second in preventing costly synthesis of T3SS3 apparatus components that are no longer required. Given the critical role of T3SS3 and T6SS1 in causing disease, BsaN/BicA could be considered a central regulator of B. pseudomallei mammalian virulence. Virulence studies in mice support this notion, since the ΔbsaN mutant was unable to cause disease [8] in contrast to the ΔbspR mutant, which produced a more chronic infection in mice compared to wildtype bacteria [14].
In addition to loci associated with T3SS3 and T6SS1, 41 other genes with potential roles in virulence were also found by RNAseq to be positively regulated by BsaN, most notably the bimBCAD intracellular motility operon and tssM. Regulation of bimA has been shown to be through virAG[8], explaining why no BsaN motif was identified for the operon. While bimA encodes an autotransporter protein that nucleates and polymerizes host cell actin to facilitate intracellular motility and cell-cell spread by the bacteria [36], the functions of the other loci in the bim operon are unknown. TssM has been shown to suppress host NFκB and Type I interferon pathways [31]. TssM is expressed and secreted inside cells following infection with B. mallei[29], however, secretion occurs independently of T3SS3 and T6SS1 [31]. BsaN was also found to activate expression of a putative non-ribosomal peptide synthase (NRPS)/polyketide synthase (PKS) biosynthesis locus. The diversity of polyketides, PKSs and NRPS/PKS hybrid systems was recently reviewed by Hertweck [37]. The B. pseudomallei locus is similar in gene content to that of a recently described plasmid encoded NRPS/PKS system in the marine bacterium Alteromonas macleodii, which was suggested to produce a bleomycin-related antibiotic Unlike A. macleodii, the gene encoding the putative bleomycin-family resistance protein (BPSL2883) is not co-localized with the NRPS/PKS gene cluster, although they are similarly regulated by BsaN (Table 1).
BsaN is homologous to the Salmonella typhimurium InvF, Shigella flexneri MxiE and the Yersinia enterocholitica YsaB transcriptional regulators [38]–[40]. All belong to the AraC/XylS family of transcriptional regulators, which act in complex with a chaperone to activate their respective T3SS genes. The chaperones not only serve as cognate partners to the transcriptional activators but also pair with T3SS translocase proteins, which are secreted into the host membrane to facilitate the injection of effector proteins [41]. We currently, have no understanding of the timed mechanism that frees BicA and allows it to partner with BsaN. The S. typhimurium chaperone SicA was shown to partition the translocase SipB and SipC, and it is sequestered by SipB [42]. Once apparatus assembly is complete, translocases are secreted and SicA is free to complex and thus activate InvF. The InvF-SicA split feedback regulatory loop, which includes positive autoregulation of invF, is conserved in Y. enterocholitica[40]. However, in S. flexneri MxiE-dependent activity is inhibited via sequestration by the T3SS substrate OspD1 when the apparatus is inactive [43]. Only when OspD1 is secreted, can MxiE partner with its chaperone IpgC to activated transcription of effector genes.
Regulation by BsaN-BicA is distinct from the previously described systems. The designation of BsaN-BicA as a dual-function regulatory protein complex is illustrated by its role in activating T3SS effector and accessory genes while repressing the system’s structural and secretion components as summarized in Figure 7. BsaN was also found to suppress the transcription of 51 additional genes in the B. pseudomallei genome including those belonging to the fla1 flagellar and chemotaxis locus on chromosome 1 (Figure 1E). Fla1 is the sole flagellar system in Southeast Asian B. pseudomallei strains such as KHW, in contrast to Australian B. pseudomallei isolates which possess a complete second system encoded on chromosome 2 (Fla2) [9],[44]. The conserved fla1 locus encodes polar flagella and was shown to be responsible for swimming in liquid medium and swarming in soft agar, but played no role in intracellular motility following infection [9]. Moreover, we were intrigued to find that BsaN suppresses a second PKS/NRPS cluster (BPSS0130, BPSS0303-BPSS0311, BPSS0328-BPSS0339) (Table 2), where almost identical homologs were identified in B. mallei and B. thailandensis by Biggins et al. and shown to produce an iron-chelating siderophore called malleilactone [45]. Disruption of the MAL cluster in B. thailandensis reduced lethality following infection of C. elegans, and purified malleilactone was toxic to mammalian cells at micromolar concentrations. How the function of MAL fits within an overall regulatory framework that promotes virulence is not clear, although it is conceivable that BsaN-mediated suppression of MAL reduces the production of toxic products during infection, thereby promoting long term survival within eukaryotic hosts. Alternatively, malleilactone itself may regulate virulence factor production similar to that reported for the P. aeruginosa siderophore pyoverdine [46].

Diagram of BsaN regulon. The BsaN regulon is shown as part of a regulatory network, which is superseded by BprP activating transcriptions of T3SS3 apparatus genes (blue) including bsaN. The bicA gene is likely initially transcribed via read through of apparatus genes. BsaN-BicA function as a complex to activate T3SS3 translocon (purple), effector (yellow), accessory (grey) and regulatory (red) genes. Transcriptional activation is indicated by green arrows. BsaN-BicA also activate virAG, which in turn activates the bimA motility genes and the T6SS1 locus. BprC activates the T6SS1 tssAB apparatus genes. BsaN-BicA also activate a non-ribosomal polyketide synthesis locus and several metabolic genes. BsaN-repressed genes as indicated by red, blunted lines include T3SS3 apparatus genes and flagellar motility genes. Only genes which have been validated by qRT-PCR are shown.
Until recently, BopA and BopE were the only two known T3SS effector proteins in B. pseudomallei. The dearth of effectors is surprising when compared to other intracellular pathogens such as Shigella and Salmonella that are known to possess numerous effectors. We have independently identified BopC (BPSS1516) as a new T3SS3 effector based on its regulatory control by BsaN/BicA. bopC is transcribed in an operon encoding its chaperone (BPSS1517) and a transposase (BPSS1518) that are also activated by BsaN/BicA. Incidentally, we had previously predicted by a genome-wide screen that BPSS1516 would encode a T3SS effector based on genomic colocalization with T3SS chaperones [47]. The BsaN regulatory motif we found in the promoters of the effectors was also recently reported to be associated with T3SS3 in a condition-dependent transcriptome study [48]. Of the T3SS3-linked effector proteins; BopA, BopC and BopE, our results suggest that BopA is the most critical for promoting cellular infection, consistent with prior studies linking BopA to intracellular survival of B. pseudomallei and B. mallei[16],[17],[49]. No cellular phenotype was evident following infection with ΔbopC or ΔbopE deletion mutants, and the ΔbopACE triple effector mutant was indistinguishable from the ΔbopA single deletion strain. As with bopE and bopC, no roles were observed for the BsaN-regulated effector candidate loci BPSS1513-1514 in cell-based virulence assays. BPSS1513 encodes a hypothetical protein and BPSS1514 is annotated as folE, a predicted GTP cyclohydrolase. Based on their genomic organization, the transcription of these loci is likely driven from the promoter upstream of BPSS1512 tssM. The secretion of HA-tagged BPSS1513 was not detected in in vitro secretion assays, although it is possible that the epitope tag could have interfered with secretion of BPSS1513, or that the assay was not performed at conditions permissive for secretion. It is intriguing why these three genes are placed under BsaN/BicA regulation by the bacterium. One possibility could be that they are important under specific stress conditions or during chronic infection.
Conclusions
Elucidating the scope of the BsaN regulon significantly enhances our understanding of B. pseudomallei pathogenic mechanisms. BsaN orchestrates the temporal and spatial expression of virulence determinants during progression through the intracellular lifecycle, promoting endosome escape and possibly evasion of autophagy through activation of T3SS3 effector loci, facilitating cell-cell spread by activation of T6SS1 and the bim intracellular motility loci, and suppressing cellular immunity via the action of the TssM ubiquitin hydrolase. BsaN also suppresses other loci that are potentially counterproductive following intracellular localization, such as the fla1 flagellar motility and chemotaxis locus, which could lead to activation of cellular immunity pathways through PAMP recognition. It is likely that the BsaN regulon and other virulence determinants that promote pathogenesis in higher mammals have been shaped primarily as a result of interactions with free-living protozoa, similar to what is believed to be the case for L. pneumophila[50]. Indeed, many of the same BsaN-regulated systems, namely T3SS and T6SS, are thought to act as “anti-predation determinants” that facilitate endosome escape and promote survival within bacteriovorus amoebae by manipulating eukaryotic pathways that are conserved from protists to humans [3]. The dual regulatory roles of BsaN – that of an activator and a suppressor – indicate that it is a key node in a regulatory program that successfully enables an environmental saprophyte to transition from the soil to surviving intracellularly.
Methods
Bacterial strains and culture conditions
Bacterial strains are listed in Table 3. Plasmids are listed in Table 4 and Additional file 1: Table S2. The B. pseudomallei wild-type strains used in this study are clinical isolates KHW. Plasmids were introduced into E. coli DH5α and S17-1 [51] strains by electro- or chemical-transformation. Plasmids were transferred into B. pseudomallei by conjugation from E. coli S17-1 on membrane filters. E. coli donors and B. pseudomallei recipients were first mixed on filters and incubated at 37°C on non-selective Luria-Bertani (LB) agar for 3 hours before transferring the filters onto selective media. In our RNA isolation for transcriptome analysis and qRT-PCR, B. pseudomallei wild-type and mutant strains were cultured in acidic (pH 5.0) RPMI medium containing 10% fetal bovine serum at 37°C for 4 hours, when bacteria were in their mid exponential growth phase (OD600 ~ 1.0). Acidification results in higher T3SS3 expression without affecting cell growth.
Bacterial mutant construction
B. pseudomallei gene deletions were generated by allelic exchange. Approximately 1 kb fragments upstream and downstream of the target gene were amplified from genomic DNA and cloned into pK18mobsacB vector [52] simultaneously using In-Fusion PCR cloning kit (Clontech). The plasmids were introduced into B. pseudomallei strains by conjugation. Homologous recombination was then selected for by growing bacteria in LB + 15% sucrose to counter select the sacB gene in the pK18mobsacB plasmid backbone. Successful double cross-over clones were screened by colony PCR from kanamycin sensitive colonies.
Activation of potential promoters by regulators
The ability of regulators to directly activate the expression of promoters was examined in E. coli DH5α as described previously [8]. Briefly, upstream regions of B. pseudomallei genes encompassing at least 100 bp of non-coding sequence upstream of the start codon were amplified from KHW genomic DNA and fused to the lacZ gene in pRW50 or pRW50mob to generate transcriptional fusions (Table 4). Coding sequences of regulators were amplified from KHW genomic DNA and cloned into the arabinose-inducible expression vector pMLBAD. The lacZ fusion plasmid and arabinose-inducible regulator plasmid were introduced into the E. coli DH5α. β-galactosidase activities arising from the expression of promoter-lacZ fusions were assessed. β-Galactosidase assays were performed and values were calculated as previously described [53].
Transcriptome analysis by RNAseq
Total RNA was extracted from three independently grown bacterial cultures that were combined at equal cell density in their exponential growth phase and quick frozen in dry ice-ethanol slurry. Approximately 2 × 109 ice cold cells were centrifuged at 3000 × g for 45 sec and 4°C and RNA was isolated from cell pellets using the RiboPure™-Bacteria Kit (Ambion). Stable RNAs were removed from 10 μg RNA using the MICROBExpress kit from Ambion. Absence of genomic DNA contamination was confirmed by PCR. Paired-end libraries for Illumina sequencing [54] were prepared using the TruSeq RNA sample preparation kit version 2.0 (Illumina) according to manufacturer's High Sample (HS) protocol albeit omitting the initial poly A selection step. Libraries were generated from 2 technical replicates using 350–500 ng enriched RNA from wildtype and ΔbsaN mutant strains as the starting material. Library preparation and sequencing was done by the UCLA Neuroscience Genomics Core (UNGC). Reads were aligned to chromosomes I and II of B. pseudomallei KHW (also called BP22) (RefSeq identification numbers NZ_CM001156.1 and NZ_CM001157.1) and B. pseudomallei K96243 (RefSeq identification numbers NC_006350.1 and NC_006351.1) as the annotated reference genome. The number of reads aligning to each genomic position on each strand was calculated and normalized using RPKM ([reads/kb of gene]/[million reads aligning to genome]). Differentially expressed genes identified by the log2 ratio of the differential between the wildtype and ΔbsaN RPKMs. Only, genes with a Δlog2 value of >1.5 and < −1.5 corresponding to 3-fold up or down regulated genes with an adjusted p value (padj) of <0.01 were considered for this study.
Measurement of B. pseudomallei gene expression by qRT- PCR
Expression of activated genes was confirmed by qRT-PCR of RNA prepared from bacteria grown in acidified RPMI. Gene repression was difficult to observe under these conditions; RNA for qRT-PCR analysis was therefore prepared from infected RAW264.7 cells using the following procedure: RAW264.7 cells (5 × 105 cells/well) were seeded and grown overnight in DMEM medium in 12 well plates. RAW264.7 cells were transferred to RPMI medium prior to infection and infected at MOI of 100:1. Bacterial RNA was isolated from infected RAW264.7 cells 4 hours post infection using TRIzol and PureLink RNA mini-kit (Invitrogen). cDNA was synthesized using 1 μg of RNA and the High Capacity Reverse Transcription Reagent Kit (Applied Biosystems). Transcripts were quantified using GoTaq qPCR Master Mix (Promega) in a BioRad iQ5 machine. Real-time PCR primers are listed in Additional file 1: Table S4. Relative RNA level of a particular gene in mutant strains was normalized to that of wild type using the 2−ΔΔCt method with 16S rRNA or recA as reference gene [55].
Mapping transcriptional start sites
The transcriptional start (+1) sites of the promoters were mapped by RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) [56]. The RLM-RACE was performed using GeneRacer Kit (Invitrogen) according to manufacturer’s instructions. The B. pseudomallei RNA was isolated as described previously [14].
Sequence motif predication and database search of motifs in the B. pseudomallei KHW genome
150 base pairs of nucleotide sequence upstream of the transcriptional starts of each gene was submitted to the bioinformatics tool – MEME (http://meme.nbcr.net/meme/cgi-bin/meme.cgi) for prediction of DNA motifs [57]. The motif with the highest statistical significance (lowest E-value) was chosen and its data – in Position-Specific Probability Matrix format was submitted to MAST (http://meme.nbcr.net/meme/cgi-bin/mast.cgi) to search for the best matching positions in the upstream sequences of B. pseudomallei KHW genes [58].
Statistical analysis
Results were presented as mean ± standard deviation. Student’s t-test was used to find the significant differences between the means, defined as when p < 0.05 (*) and p < 0.01 (**).
Additional file
References
Galyov EE, Brett PJ, DeShazer D: Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu Rev Microbiol. 2010, 64: 495-517. 10.1146/annurev.micro.112408.134030.
Wiersinga WJ, van der Poll T, White NJ, Day NP, Peacock SJ: Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat Rev Microbiol. 2006, 4 (4): 272-282. 10.1038/nrmicro1385.
Hasselbring BM, Patel MK, Schell MA: Dictyostelium discoideum as a model system for identification of Burkholderia pseudomallei virulence factors. Infect Immun. 2011, 79 (5): 2079-2088. 10.1128/IAI.01233-10.
Inglis TJ, Rigby P, Robertson TA, Dutton NS, Henderson M, Chang BJ: Interaction between Burkholderia pseudomallei and Acanthamoeba species results in coiling phagocytosis, endamebic bacterial survival, and escape. Infect Immun. 2000, 68 (3): 1681-1686. 10.1128/IAI.68.3.1681-1686.2000.
Lee YH, Chen Y, Ouyang X, Gan YH: Identification of tomato plant as a novel host model for Burkholderia pseudomallei. BMC Microbiol. 2010, 10: 28-10.1186/1471-2180-10-28.
Kaestli M, Schmid M, Mayo M, Rothballer M, Harrington G, Richardson L, Hill A, Hill J, Tuanyok A, Keim P, Hartmann A, Currie BJ: Out of the ground: aerial and exotic habitats of the melioidosis bacterium Burkholderia pseudomallei in grasses in Australia. Environ Microbiol. 2012, 14 (8): 2058-2070. 10.1111/j.1462-2920.2011.02671.x.
Burtnick MN, Brett PJ, Harding SV, Ngugi SA, Ribot WJ, Chantratita N, Scorpio A, Milne TS, Dean RE, Fritz DL, Peacock SJ, Prior JL, Atkins TP, Deshazer D: The Cluster 1 Type VI Secretion System is a Major Virulence Determinant in Burkholderia pseudomallei. Infect Immun. 2011, 79 (4): 1512-1525. 10.1128/IAI.01218-10.
Chen Y, Wong J, Sun GW, Liu Y, Tan GY, Gan YH: Regulation of type VI secretion system during Burkholderia pseudomallei infection. Infect Immun. 2011, 79 (8): 3064-3073. 10.1128/IAI.05148-11.
French CT, Toesca IJ, Wu TH, Teslaa T, Beaty SM, Wong W, Liu M, Schröder I, Chiou PY, Teitell MA, Miller JF: Dissection of the Burkholderia intracellular life cycle using a photothermal nanoblade. Proc Natl Acad Sci U S A. 2011, 108 (29): 12095-12100. 10.1073/pnas.1107183108.
Stevens MP, Wood MW, Taylor LA, Monaghan P, Hawes P, Jones PW, Wallis TS, Galyov EE: An Inv/Mxi-Spa-like type III protein secretion system in Burkholderia pseudomallei modulates intracellular behaviour of the pathogen. Mol Microbiol. 2002, 46 (3): 649-659. 10.1046/j.1365-2958.2002.03190.x.
Sun GW, Lu J, Pervaiz S, Cao WP, Gan YH: Caspase-1 dependent macrophage death induced by Burkholderia pseudomallei. Cell Microbiol. 2005, 7 (10): 1447-1458. 10.1111/j.1462-5822.2005.00569.x.
Stevens MP, Haque A, Atkins T, Hill J, Wood MW, Easton A, Nelson M, Underwood-Fowler C, Titball RW, Bancroft GJ, Galyov EE: Attenuated virulence and protective efficacy of a Burkholderia pseudomallei bsa type III secretion mutant in murine models of melioidosis. Microbiology. 2004, 150 (Pt 8): 2669-2676. 10.1099/mic.0.27146-0.
Warawa J, Woods DE: Type III secretion system cluster 3 is required for maximal virulence of Burkholderia pseudomallei in a hamster infection model. FEMS Microbiol Lett. 2005, 242 (1): 101-108. 10.1016/j.femsle.2004.10.045.
Sun GW, Chen Y, Liu Y, Tan GY, Ong C, Tan P, Gan YH: Identification of a regulatory cascade controlling Type III Secretion System 3 gene expression in Burkholderia pseudomallei. Mol Microbiol. 2010, 76 (3): 677-689. 10.1111/j.1365-2958.2010.07124.x.
Stevens MP, Friebel A, Taylor LA, Wood MW, Brown PJ, Hardt WD, Galyov EE: A Burkholderia pseudomallei type III secreted protein, BopE, facilitates bacterial invasion of epithelial cells and exhibits guanine nucleotide exchange factor activity. J Bacteriol. 2003, 185 (16): 4992-4996. 10.1128/JB.185.16.4992-4996.2003.
Cullinane M, Gong L, Li X, Lazar-Adler N, Tra T, Wolvetang E, Prescott M, Boyce JD, Devenish RJ, Adler B: Stimulation of autophagy suppresses the intracellular survival of Burkholderia pseudomallei in mammalian cell lines. Autophagy. 2008, 4 (6): 744-753. 10.4161/auto.6246.
Gong L, Cullinane M, Treerat P, Ramm G, Prescott M, Adler B, Boyce JD, Devenish RJ: The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS One. 2011, 6 (3): e17852-10.1371/journal.pone.0017852.
Muangman S, Korbsrisate S, Muangsombut V, Srinon V, Adler NL, Schroeder GN, Frankel G, Galyov EE: BopC is a type III secreted effector protein of Burkholderia pseudomallei. FEMS Microbiol Lett. 2011, 323 (1): 75-82. 10.1111/j.1574-6968.2011.02359.x.
Srinon V, Muangman S, Imyaem N, Muangsombut V, Lazar Adler NR, Galyov EE, Korbsrisate S: Comparative assessment of the intracellular survival of the Burkholderia pseudomallei bopC mutant. J Microbiol. 2013, 51 (4): 522-526. 10.1007/s12275-013-2557-3.
Liu B, Koo GC, Yap EH, Chua KL, Gan YH: Model of differential susceptibility to mucosal Burkholderia pseudomallei infection. Infect Immun. 2002, 70 (2): 504-511. 10.1128/IAI.70.2.504-511.2002.
Biggins JB, Liu X, Feng Z, Brady SF: Metabolites from the induced expression of cryptic single operons found in the genome of Burkholderia pseudomallei. J Am Chem Soc. 2011, 133 (6): 1638-1641. 10.1021/ja1087369.
Tang H, Braun TF, Blair DF: Motility protein complexes in the bacterial flagellar motor. J Mol Biol. 1996, 261 (2): 209-221. 10.1006/jmbi.1996.0453.
Cotter PA, Melville SB, Albrecht JA, Gunsalus RP: Aerobic regulation of cytochrome d oxidase (cydAB) operon expression in Escherichia coli: roles of Fnr and ArcA in repression and activation. Mol Microbiol. 1997, 25 (3): 605-615. 10.1046/j.1365-2958.1997.5031860.x.
Lefebre MD, Valvano MA: Construction and evaluation of plasmid vectors optimized for constitutive and regulated gene expression in Burkholderia cepacia complex isolates. Appl Environ Microbiol. 2002, 68 (12): 5956-5964. 10.1128/AEM.68.12.5956-5964.2002.
Sambrook J, Maniatis T, Fritsch EF: Molecular cloning : a laboratory manual. 1989, Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y
Lodge J, Fear J, Busby S, Gunasekaran P, Kamini NR: Broad host range plasmids carrying the Escherichia coli lactose and galactose operons. FEMS Microbiol Lett. 1992, 74 (2–3): 271-276. 10.1111/j.1574-6968.1992.tb05378.x.
DeShazer D, Brett PJ, Carlyon R, Woods DE: Mutagenesis of Burkholderia pseudomallei with Tn5-OT182: isolation of motility mutants and molecular characterization of the flagellin structural gene. J Bacteriol. 1997, 179 (7): 2116-2125.
Holden MT, Titball RW, Peacock SJ, Cerdeño-Tárraga AM, Atkins T, Crossman LC, Pitt T, Churcher C, Mungall K, Bentley SD, Sebaihia M, Thomson NR, Bason N, Beacham IR, Brooks K, Brown KA, Brown NF, Challis GL, Cherevach I, Chillingworth T, Cronin A, Crossett B, Davis P, DeShazer D, Feltwell T, Fraser A, Hance Z, Hauser H, Holroyd S, Jagels K: Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc Natl Acad Sci U S A. 2004, 101 (39): 14240-14245. 10.1073/pnas.0403302101.
Shanks J, Burtnick MN, Brett PJ, Waag DM, Spurgers KB, Ribot WJ, Schell MA, Panchal RG, Gherardini FC, Wilkinson KD, Deshazer D: Burkholderia mallei tssM encodes a putative deubiquitinase that is secreted and expressed inside infected RAW 264.7 murine macrophages. Infect Immun. 2009, 77 (4): 1636-1648. 10.1128/IAI.01339-08.
Teh BE, French CT, Chen Y, Chen IG, Wu TH, Sagullo E, Chiou PY, Teitell MA, Miller JF, Gan YH: Type three secretion system-mediated escape of Burkholderia pseudomallei into the host cytosol is critical for the activation of NFkappaB. BMC Microbiol. 2014, 14: 115-10.1186/1471-2180-14-115.
Tan KS, Chen Y, Lim YC, Tan GY, Liu Y, Lim YT, Macary P, Gan YH: Suppression of host innate immune response by Burkholderia pseudomallei through the virulence factor TssM. J Immunol. 2010, 184 (9): 5160-5171. 10.4049/jimmunol.0902663.
Raymond B, Young JC, Pallett M, Endres RG, Clements A, Frankel G: Subversion of trafficking, apoptosis, and innate immunity by type III secretion system effectors. Trends Microbiol. 2013, 21 (8): 430-441. 10.1016/j.tim.2013.06.008.
Jani AJ, Cotter PA: Type VI secretion: not just for pathogenesis anymore. Cell Host Microbe. 2010, 8 (1): 2-6. 10.1016/j.chom.2010.06.012.
Wong KT, Puthucheary SD, Vadivelu J: The histopathology of human melioidosis. Histopathology. 1995, 26 (1): 51-55. 10.1111/j.1365-2559.1995.tb00620.x.
Cascales E, Cambillau C: Structural biology of type VI secretion systems. Philos Trans R Soc Lond B Biol Sci. 2012, 367 (1592): 1102-1111. 10.1098/rstb.2011.0209.
Stevens MP, Stevens JM, Jeng RL, Taylor LA, Wood MW, Hawes P, Monaghan P, Welch MD, Galyov EE: Identification of a bacterial factor required for actin-based motility of Burkholderia pseudomallei. Mol Microbiol. 2005, 56 (1): 40-53. 10.1111/j.1365-2958.2004.04528.x.
Hertweck C: The biosynthetic logic of polyketide diversity. Angew Chem Int Ed Engl. 2009, 48 (26): 4688-4716. 10.1002/anie.200806121.
Darwin KH, Miller VL: Type III secretion chaperone-dependent regulation: activation of virulence genes by SicA and InvF in Salmonella typhimurium. EMBO J. 2001, 20 (8): 1850-1862. 10.1093/emboj/20.8.1850.
Kane CD, Schuch R, Day WA, Maurelli AT: MxiE regulates intracellular expression of factors secreted by the Shigella flexneri 2a type III secretion system. J Bacteriol. 2002, 184 (16): 4409-4419. 10.1128/JB.184.16.4409-4419.2002.
Walker KA, Miller VL: Regulation of the Ysa type III secretion system of Yersinia enterocolitica by YsaE/SycB and YsrS/YsrR. J Bacteriol. 2004, 186 (13): 4056-4066. 10.1128/JB.186.13.4056-4066.2004.
Deane JE, Abrusci P, Johnson S, Lea SM: Timing is everything: the regulation of type III secretion. Cell Mol Life Sci. 2010, 67 (7): 1065-1075. 10.1007/s00018-009-0230-0.
Tucker SC, Galan JE: Complex function for SicA, a Salmonella enterica serovar typhimurium type III secretion-associated chaperone. J Bacteriol. 2000, 182 (8): 2262-2268. 10.1128/JB.182.8.2262-2268.2000.
Parsot C, Ageron E, Penno C, Mavris M, Jamoussi K, d'Hauteville H, Sansonetti P, Demers B: A secreted anti-activator, OspD1, and its chaperone, Spa15, are involved in the control of transcription by the type III secretion apparatus activity in Shigella flexneri. Mol Microbiol. 2005, 56 (6): 1627-1635. 10.1111/j.1365-2958.2005.04645.x.
Tuanyok A, Auerbach RK, Brettin TS, Bruce DC, Munk AC, Detter JC, Pearson T, Hornstra H, Sermswan RW, Wuthiekanun V, Peacock SJ, Currie BJ, Keim P, Wagner DM: A horizontal gene transfer event defines two distinct groups within Burkholderia pseudomallei that have dissimilar geographic distributions. J Bacteriol. 2007, 189 (24): 9044-9049. 10.1128/JB.01264-07.
Biggins JB, Ternei MA, Brady SF: Malleilactone, a polyketide synthase-derived virulence factor encoded by the cryptic secondary metabolome of Burkholderia pseudomallei group pathogens. J Am Chem Soc. 2012, 134 (32): 13192-13195. 10.1021/ja3052156.
Lamont IL, Beare PA, Ochsner U, Vasil AI, Vasil ML: Siderophore-mediated signaling regulates virulence factor production in Pseudomonasaeruginosa. Proc Natl Acad Sci U S A. 2002, 99 (10): 7072-7077. 10.1073/pnas.092016999.
Panina EM, Mattoo S, Griffith N, Kozak NA, Yuk MH, Miller JF: A genome-wide screen identifies a Bordetella type III secretion effector and candidate effectors in other species. Mol Microbiol. 2005, 58 (1): 267-279. 10.1111/j.1365-2958.2005.04823.x.
Ooi WF, Ong C, Nandi T, Kreisberg JF, Chua HH, Sun G, Chen Y, Mueller C, Conejero L, Eshaghi M, Ang RM, Liu J, Sobral BW, Korbsrisate S, Gan YH, Titball RW, Bancroft GJ, Valade E, Tan P: The condition-dependent transcriptional landscape of Burkholderia pseudomallei. PLoS Genet. 2013, 9 (9): e1003795-10.1371/journal.pgen.1003795.
Whitlock GC, Estes DM, Young GM, Young B, Torres AG: Construction of a reporter system to study Burkholderia mallei type III secretion and identification of the BopA effector protein function in intracellular survival. Trans R Soc Trop Med Hyg. 2008, 102 (Suppl 1): S127-133. 10.1016/S0035-9203(08)70029-4.
Escoll P, Rolando M, Gomez-Valero L, Buchrieser C: From Amoeba to Macrophages: Exploring the Molecular Mechanisms of Legionella pneumophila Infection in Both Hosts. Curr Top Microbiol Immunol. 2013, 376: 1-34.
Simon R, Priefer U, Pühler A: A broad range mobilization system for in vitro genetic engineering: Transposon mutagenesis in Gram-negative bacteria. Bio/Technology. 1983, 1: 784-791. 10.1038/nbt1183-784.
Schafer A, Tauch A, Jager W, Kalinowski J, Thierbach G, Puhler A: Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994, 145 (1): 69-73. 10.1016/0378-1119(94)90324-7.
Zhang X, Bremer H: Control of the Escherichia coli rrnB P1 promoter strength by ppGpp. J Biol Chem. 1995, 270 (19): 11181-11189. 10.1074/jbc.270.19.11181.
Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A: Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods. 2010, 7 (9): 709-715. 10.1038/nmeth.1491.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001, 25 (4): 402-408. 10.1006/meth.2001.1262.
Bensing BA, Meyer BJ, Dunny GM: Sensitive detection of bacterial transcription initiation sites and differentiation from RNA processing sites in the pheromone-induced plasmid transfer system of Enterococcus faecalis. Proc Natl Acad Sci U S A. 1996, 93 (15): 7794-7799. 10.1073/pnas.93.15.7794.
Bailey TL, Elkan C: Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994, 2: 28-36.
Bailey TL, Gribskov M: Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998, 14 (1): 48-54. 10.1093/bioinformatics/14.1.48.
Acknowledgements
We thank M. A. Valvano (University of Western Ontario) for pMLBAD plasmid, S.J. Busby (University of Birmingham) for pRW50 plasmid, S. Korbsrisate (Mahidol University) for BopC antibody, and M.P. Stevens (University of Edinburgh) for BopE antibody. This work is supported by grants T208A3105 from the Ministry of Education to YHG, NMRC/1221/2009 from the National Medical Research Council to YHG, an award from the Pacific Southwest Regional Center of Excellence in Biodefense and Emerging Infectious Diseases (NIH U54 A1065359) to JFM, and grant HDTRA1-11-1-0003 from the Defense Threat Reduction Agency to JFM. We would like to thank Isabelle Chen for her technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no competing interests.
Authors’ contributions
YC, IS and YHG designed the experiments. YC, IS, CTF, XJY, BET and IJT performed the experiments. YC, IS, CTF, AJ and YHG analyzed the results. YC and YHG conceived the study and together with IS and JFM wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12866_2014_206_MOESM1_ESM.docx
Additional file 1:Materials and Methods. Table S1. Summary of Illumina sequencing. Table S2. β-galactosidase activities in E coli DH5α strain containing transcriptional promoter-lacZ fusions and arabinose-inducible bsaN and bicA or empty vector. Table S3. List of additional plasmids used in this study. Table S4. List of Real-Time PCR primers for this study. Figure S1. Secretion of BopC. KHW and ΔbsaM mutant were grown in acidic LB broth for 3 hours. Total protein from the bacterial culture supernatant was precipitated and protein concentration was normalized with respect to the optical density (OD600) of the bacterial cultures. Proteins on membranes were probed with rabbit polyclonal antibodies to BopC and BopE. Figure S2. (A) Intracellular replication of B. pseudomallei KHW and mutants in RAW264.7 cells. Cells were infected at an MOI of 0.1:1. Intracellular bacterial loads were quantified at 2 and 8 h post infection by plate counting. (B) Cytotoxicity of B. pseudomallei KHW and mutants against RAW264.7 cells. Cells were infected at an MOI of 100:1. Cytotoxicity was quantified at 8 h post infection by LDH release assay. *p < 0.05. Figure S3. Secretion and function of BsaN controlled proteins. A. Secretion of BPSS1513 in strain KHW. Proteins were separated on 12% polyacrylamide gels, transferred to PVDF membranes and probed with a mouse monoclonal antibody to HA or rabbit polyclonal antibody to BopE. P: pellet; S: supernatant. B. Intracellular replication of B. pseudomallei KHW and Δ(BPSS1513-folE) mutant in RAW264.7 cells at 2 h and 8 h (MOI of 10:1) or C. 2 h and 24 h after infection at an MOI of 0.1:1. Intracellular bacterial loads were quantified by plate counting. D. Cytotoxicity of B. pseudomallei KHW and Δ(BPSS1513-folE) mutant against RAW264.7 cells. Cells were infected at an MOI of 100:1. Cytotoxicity was quantified at 8 h post infection by LDH release assay. E. MNGC formation of cells infected with B. pseudomallei wild-type (WT) strain KHW and F. Δ(BPSS1513-1514) mutant at an MOI of 10:1. (DOCX 1000 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Chen, Y., Schröder, I., French, C.T. et al. Characterization and analysis of the Burkholderia pseudomallei BsaN virulence regulon. BMC Microbiol 14, 206 (2014). https://doi.org/10.1186/s12866-014-0206-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-014-0206-6