
Abstract
Objective
This study was aimed to identify the biomarkers for diagnosis and reveal the immune microenvironment changes in ankylosing spondylitis (AS).
Methods
GSE73754 was downloaded for the co-expression network construction and immune cell analyses. Flow cytometric analysis was performed to validate the results of bioinformatics analysis. Gene set enrichment analysis (GSEA) was performed to investigate the potential biological characteristic between different phenotypes. Pearson correlation analysis between the hub genes and the xCell score of immune cell types was performed.
Results
Signal transducer and activator of transcription 3 (STAT3) and Spi-1 proto-oncogene (SPI1) was identified as the hub genes in the datasets GSE73754. And the t-test showed that the expression level of STAT3 and SPI1 in the GSE73754 was significantly higher in AS and human leukocyte antigen (HLA)-B27(+) groups. Flow cytometric analysis showed that natural killer T cells (NKT) cells were upregulated, while Th1 cells were down-regulated in AS, which was consistent with the results obtained from bioinformatics analysis. STAT3 and SPI1 was correlated with the NKT cells and Th1 cells.
Conclusion
STAT3 and SPI1 may be a key cytokine receptor in disease progression in AS.
Similar content being viewed by others

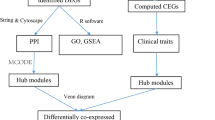

Introduction
Ankylosing spondylitis (AS), a chronic inflammatory autoimmune disease, is a global disease diagnosed by millions of people every year, and it mainly occurred in young adult males [1]. AS mostly involves sacroiliac joints and axial skeleton and finally resulting in impairments of structure and function [2]. AS is caused by the loss of tolerance to autoantigens and autoantibodies' development because of the effectors of innate and adaptive immunity, which ultimately results in tissue inflammation and damage [3]. AS's pathogenesis is multifactorial, involving multiple cell types, cellular processes, and molecular mediators and regulators [4, 5]. Studies show that the major histocompatibility complex region (MHC) is closely related to AS, and about 95% AS patients are born with a gene named HLA-B27 [6]. However, HLA-B27 predisposition fails to fully explain the cause of the disease, and this leads to a strong effort to identify additional predisposing factors.
Dysregulation of the immune cells conferred risk for the pathogenesis of human autoimmune diseases including AS [7]. The balance between 1 helper T (Th1) and Th2 cells regulates the choice between inflammatory and antibody-mediated immune responses [8]. Those antibodies contain the autoantibody. The balance between Th1 and Th2 cells may also related to AS. A previous study suggested that Th17 cells and NKT cells secrete IL-17, which is a crucial pathologic cytokine in AS pathogenesis [9]. However, which genes lead to immune system dysregulation in AS also remained unknown. Therefore, there is an urgent need to predict the pathogenesis of AS.
Heterotopic ossification is an important factor in spinal fusion in AS. Bone formation requires critical osteogenic molecules and pathways, inflammation-induced activation of osteogenic signaling pathways is considered a logical link between inflammation and new bone formation. Wnt, MAPK, JAK/STAT signaling pathways have been previously investigated and shown to be involved in spinal fusion in patients with AS [10,11,12]. There are many cytokines being identified that regulate osteoblasts/osteoclasts differentiation. Moreover, immune cells can secrete cytokines which play a key role in osteoblasts/osteoclasts differentiation. Thus, in the present study, we utilized several bioinformatics approaches to further investigate the molecule mechanism of AS. How the key genes affect immune cells that leads to heterotopic ossification in AS. That may help develop new therapies that treat AS by exploring the complex mechanisms of immune responses.
In the present study, STAT3 and SPI1, which were identified as hub genes, could work as biomarkers for AS diagnosis. Secondly, we demonstrated STAT3 and SPI1 were involved in the activation, proliferation, and differentiation of Th1 cells and NKT cells in AS. STAT3 and SPI1 were involved in heterotopic ossification in AS. Additionally, the balance between Th1 cells and NKT cells may related to the osteogenesis and osteolysis in AS. STAT3 and SPI1 may be the key genes that lead to the dysregulation of the immune system and contributing to the pathogenesis of AS.
Results
Identification of stable modules associated with AS
WGCNA was performed on the 7395 genes of 72 samples in GSE73754. A total of 68 samples remained for subsequent analysis after four outlier samples were removed (Fig. 1A). The connectivity between genes in the gene network met a scale-free network distribution with a soft threshold power of β = 6 (scale-free R2 = 0.85) (Fig. 1B, C). After merging similar clusters, fourteen modules that contained groups of genes with shown in similar patterns of connection strengths with other genes were identified (Fig. 2A). We found significant correlation between HLA-B27 (+) and “red module” in patients with AS (Fig. 2B). In addition, scatterplots of gene significance versus module membership in the red module showed that they were highly correlated (Fig. 2C, D).

Weighted gene co-expression network analysis. A Clustering of samples to detect outliers in GSE73754. B The scale-free fit index as a function of the soft-threshold power. C The mean connectivity as a function of the soft-threshold power

The results of WGCNA. A Cluster dendrogram based on the dynamic tree. Different dendrogram colors represent various modules. B Heatmap of the correlation between module eigengenes and different clinical characteristics of AS. C A scatterplot of gene significance for AS vs. module membership in the red module. D A scatterplot of gene significance for HLA-B27(+) versus module membership in the red module
Enrichment analyses and diagnosis ability of the hub genes
A total of 416 genes in red module were performed enrichment analyses. GO analysis of the genes in red module in the GSE73754 illustrated that the main biological processes (BP), cellular components (CC) and molecular functions (MF) were all related to immune response (Fig. 3A). KEGG pathway analysis to the genes in red module in the GSE73754 suggested that the genes were particularly involved in immune response and immune cell differentiation (Fig. 3B).

The GO\KEGG analysis results and PPI network for the DEGs in the red module in the GSE73754. The GO (A) and KEGG (B) analysis results for the DEGs in the red module in the GSE73754. C PPI network for the DEGs in the red module in the GSE73754. The red circle showed the position of STAT3 and SPI1. D The top five genes were arranged in descending order of “Degree”, “Betweenness”, and “Closeness” respectively
In order to screen out the hub genes from the genes in red module, all the genes in red module were uploaded to the STRING for further analysis. The PPI network were showed in Fig. 3C. Figure 3D showed the top five genes which were arranged in descending order of “Degree”, “Betweenness”, and “Closeness” respectively. STAT3 and SPI1 were identified as hub genes in the GSE73754 (Fig. 4A). Calibration curves indicated a satisfactory agreement between prediction and actual probabilities (Fig. 4B), prediction efficiency was much better especially when STAT3 and SPI1 combined. Furthermore, the AUCs of the ROC curves were 0.774 for STAT3, 0.829 for SPI1, and 0.836 for both combined (Fig. 4C). A nomogram (Fig. 4D) based on STAT3 and SPI1 was constructed for advancing clinical diagnosis of AS.

Hub genes identification. A The Venn diagram showed the hub genes in the GSE73754. B The calibration plots of different model for diagnosis. C ROC curves of hub genes’ diagnostic ability. D Nomogram for predicting the probability of AS
Relationship between STAT3 and SPI1 and clinical traits
STAT3 and SPI1 were up-regulated in the AS group (Fig. 5A). In addition, STAT3 and SPI1 were also up-regulated in the HLA-B27(+) group (Fig. 5B). Both STAT3 and SPI1 expression were up-regulated in both male and female patients with AS (Fig. 5C, D). However, there were no significant difference between male and female patients with AS (Fig. 5E, F).

The correlation of STAT3 and SPI1 expression with clinical characteristics. A The expression of STAT3 and SPI1 between AS and HCs. B The expression of STAT3 and SPI1 between HLA-B27 (−) and HLA-B27 (+). C The expression of STAT3 and SPI1 between AS and HCs in female patients. D The expression of STAT3 and SPI1 between AS and HCs in male patients. E The expression of STAT3 and SPI1 between male and female in AS patients. F The expression of STAT3 and SPI1 between male and female in all patients
Correlation between STAT3 and SPI1 and immune cells
The cell types potentially involved in AS were determined using xCell, which generates cell type enrichment scores using two datasets' gene expression data. The cell types that were significantly different between AS and HCs in the GSE73754 dataset are shown in Additional file 1: Fig. S1. We evaluate the correlation between the xCell scores of immune cells and the expression level of STAT3 and SPI1. STAT3 and SPI1 were significantly correlated with the xCell scores of NKT cells, Th1 cells, and smooth muscle in the GSE73754 (Fig. 6A, B). Flow cytometry was performed to validate the difference between NKT cells and Th1 cells between AS and HCs. NKT cells were upregulated (Fig. 6C, D), while Th1 cells were downregulated in AS (Fig. 6E, F), which was consistent with the results obtained from bioinformatics analysis.

The correlation of STAT3 and SPI1 expression with xCell score. A The correlation of the expression of STAT3 with xCell score in GSE73754. B The correlation of the expression of SPI1 with xCell score in GSE73754. C, E Dot plots of a representative AS patient and HC. D Percentage of Th1 cells (% of CD4 + IFNγ + T cells). F Percentage of NKT cells (% of CD3 + CD56 + T cells)
GSEA analysis
GSEA results on KEGG pathways revealed that most of the gene sets focused on immune-related pathways. A significant number of genes were enriched in pathways related to immunity among others. KEGG results showed that a majority of the genes were enriched in Fc-gamma R mediated phagocytosis, JAK/STAT signaling pathway, MAPK signaling pathway, VEGF receptor signaling pathway, and Wnt receptor signaling pathway in the AS group (Fig. 7A). KEGG analyses to the high STAT3/SPI1 expression and high-NKT xCell score showed the similar results in the AS group (Fig. 7B–D). KEGG results showed that most of the genes were enriched in oxidative phosphorylation, primary-immunodeficiency, proteasome, T-cell receptor signaling pathway in the high-Th1 xCell score group (Fig. 7E). The results provide clues into the in-depth biomechanism of AS and maybe a foundation for the subsequent application of AS immunotherapy.

GSEA analysis. A GSEA in the KEGG gene set between AS (n = 52) and HCs (n = 20) group in GSE73754. B–E GSEA results of the KEGG backgroung between high (n = 36) and low (n = 36) of gene expression and xCell score in GSE73754
Discussion
It has been proved that AS is a chronic autoimmunity disease characterized by inflammation in the spine and sacroiliac joints resulting in initial bone and joint erosion [16]. As we know, the onset of AS suffers from the relationship between the host genetics, the intestinal microbiome, and the immune response [17]. Autoimmune dysregulation leads to tissue inflammation and damage because of the loss of tolerance to autoantigens and autoantibodies' development. Hence, developing a better understanding of the mechanism of autoimmune dysregulation and its relationship with the key genes in AS is paramount.
In the present study, STAT3 and SPI1 was identified as the hub genes, which was up-regulated in AS. STAT3 is a member of the signal transducer and activator of transcription protein family, which are phosphorylated by the receptor associated kinases, and then form homo- or heterodimers that translocate to the cell nucleus where they act as transcription activators [18]. STAT3 mediates the expression of a variety of genes in response to cell stimuli, and thus plays a key role in many cellular processes such as cell growth, apoptosis, activation and differentiation [19]. A previous study reported that the expression of STAT3 and proteins were elevated during active RA synovitis [20]. STAT3 had also been reported associated with the pathogenesis of AS. Within the osteoblast lineage, STAT3 signaling promotes bone formation in normal physiology and in response to mechanical loading through direct signaling in osteocytes [21]. The GSEA results showed that JAK/STAT signaling pathway, MAPK signaling pathway, and Wnt receptor signaling pathway were enriched in the AS group. Additionally, a majority of the genes were also enriched in these pathways in the high STAT3 expression group. JAK/STAT, Wnt receptor, and MAPK signaling pathway were strongly associated with osteoblast differentiation and bone formation. These results demonstrated STAT3 may be involved in osteoblast differentiation and bone formation in AS patients via JAK/STAT, Wnt receptor, and MAPK signaling pathways.
In this study, STAT3 was significantly positively correlated with the xCell scores of NKT cells and negatively correlated with the xCell scores of Th1 cells. STAT3 plays a key role in activating NKT cells by targeting its downstream MHC class I-related chain (MIC) [22]. The activation of STAT3 may suppresses Th1 response during tuberculosis infection [23]. TIRC7 inhibits Th1 cells by upregulating the expression of CTLA‑4 and STAT3 in mice with acute graft‑versus‑host disease [24]. These results demonstrated that the upregulating of STAT3 may activate NKT cells and suppresses Th1 response. NKT cells could inhibit osteoclastogenesis by secreting large amounts IL‐4, which is known to be a suppressive factor for osteoclastogenesis [25]. Th1 cells has biphasic effects in the procedure of osteoclastogenesis by secreting IFN-γ and TNF-α, which can promote or inhibit the differentiation of osteoclasts [26]. As for STAT3, which was upregulated in AS patients, it can directly or indirectly induce the differentiation of osteoclasts.
SPI1 (PU.1) encodes an ETS-domain transcription factor that activates gene expression during myeloid and B-lymphoid cell development. Diseases associated with SPI1 include inflammatory diarrhea and erythroleukemia [27,28,29,30,31,32]. Among its related pathways are glucocorticoid receptor regulatory network and IL-4 signaling and its primary biological effects in different immune cell types. SPI1 have been previously shown to play important roles in the differentiation and function of osteoblasts [33]. SPI1 regulates dental pulp stem cell osteoblastic differentiation through noggin [34]. Elevated SPI1 promotes BMP2 transcription and further accumulates vascular calcification [35]. The GSEA results also showed that JAK/STAT signaling pathway, MAPK signaling pathway, and Wnt receptor signaling pathway were enriched in the high SPI1 expression group. As we just mentioned, these pathways were strongly associated with osteoblast differentiation and bone formation. There are also many studies demonstrated that up-regulated PU.1 may lead to osteoporosis by promoting osteoclast differentiation [36,37,38,39]. PU.1 may cause osteoclast differentiation and osteogenesis simultaneously in AS.
In this study, SPI1 was also significantly positively correlated with the xCell scores of NKT cells and negatively correlated with the xCell scores of Th1 cells. PU.1 involved in the transcriptional regulation of CD1D1, which plays an important role in the development and function of NKT cells regulating innate and adaptive immunity, by binding to the CD1D1 promoter [40]. Besides, PU.1 also involved in the differentiation of TH1 cells [41,42,43,44,45]. PU.1 may also play a role in heterotopic ossification and osteoporosis in AS by regulating NKT and TH1 cells differentiation.
In the present study, we demonstrated STAT3 and SPI1 may be involved in osteoblast differentiation and bone formation in AS patients via JAK/STAT, Wnt receptor, and MAPK signaling pathways. Besides, STAT3 and SPI1 may also affect heterotopic ossification and osteoporosis in AS through regulating NKT and TH1 cells differentiation. However, there are some limitations that exist in this study. We just test NKT cells and Th1 cells in peripheral blood by flow cytometry, which made our research not strict adequate and the experimental conclusion was not sufficient enough.
Conclusion
According to our analysis of the microarray datasets related to AS, STAT3 and SPI1 were identified as the hub genes, could work as diagnostic markers of AS. STAT3 and SPI1 may be involved in the NKT and Th1 cells activation, proliferation and differentiation in AS. Moreover, STAT3 and SPI1 may be involved in osteoblast differentiation and bone formation in AS patients via JAK/STAT, Wnt receptor, and MAPK signaling pathways or through regulating NKT and TH1 cells differentiation.
Materials and methods
GEO datasets
Gene expression profile data were obtained from the gene expression omnibus (GEO) (www.ncbi.nlm.nih.gov/gds), a public repository at the National Center of Biotechnology Information that contains high-throughput gene expression datasets. GSE73754, which contains 52 AS and 20 healthy controls (HCs), was downloaded for further analysis.
Weighted Correlation Network Analysis (WGCNA) network construction and module identification
We used the WGCNA (version 1.70-3) R package to construct the co-expression network. Before WGCNA, samples with obvious outlier were excluded and genes with low quality were screened and filtered. The R function pick soft-threshold was used to calculate the soft thresholding power β, to which co-expression similarity is raised to calculate adjacency. We performed hierarchical clustering to identify modules, each containing at least 50 genes. Next, we calculated the eigengene, hierarchically clustered the modules, and merged similar modules (abline = 0.25). Gene significance and module membership were calculated to relate modules to clinical characteristics. The corresponding module gene information was extracted for further analysis.
KEGG/GO enrichment analyses and hub genes identification
The genes in the red model were performed enrichment analyses using the “clusterProfiler” package (version 3.16.0) and then visualized using the ‘ggplot2’ package (version 3.3.0) in R studio. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) terms with adjusted p-value < 0.05 were considered significantly enriched. The protein–protein interaction (PPI) network was predicted using the search tool for the retrieval of interacting genes (STRING; http://string-db.org) (version 11.0) online database and was visualized using Cytoscape (version 3.6.1). The cut-off value for STRING analysis is 0.04. We used the Cytoscape plug-in APP “cytoHubba” for hub genes identification and the top 5 nodes ranked by “Degree”, “Betweenness”, and “Closeness” were enrolled in gene selection. And the overlap genes were further evaluated their diagnosis ability and relationship with clinical characteristics.
Immune and stromal cells analyzed
The ‘xCell’ package was used to calculate the cell type enrichment score (xCell scores) based on the gene expression data. Notably, the xCell tool provides 64 cell types, including lymphoid, myeloid, stromal cells, stem cells, and other cells. The difference between each cell subtype between AS and HCs was compared using the t-test method. The cut-off values for the cell analyses were p value < 0.05.
Correlation test between hub genes and immune cells
Pearson correlation analysis between SPI1 and STAT3, and the xCell score of the significant difference cell types was performed with the ‘corrplot’ package (version 0.84). Pearson correlation coefficient > 0.4 and p value < 0.05 were considered as significantly correlated.
Flow cytometry analysis
A total of 24 patients included 12 AS patients and 12 lumbar spinal stenosis patients as a control group were enrolled in this study (Table 1). All patients with AS were newly diagnosed without medicine treatment and the patients with co-diagnosis were excluded. All the AS patients were diagnosed according to the modified New York criteria (Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria) [13]. Subjects volunteering for the study had signed informed consent forms. In addition, our study was approved by the Ethics Department of the First Affiliated Hospital of Guangxi Medical University (KE-E-044) and conformed to the World Medical Association Declaration of Helsinki.
Blood samples were taken in the Department of Spine and Osteopathy Ward, the First Affiliated Hospital of Guangxi Medical University. After peripheral venous blood sampling, 5-mL samples preserved with Ethylene Diamine Tetraacetic Acid (EDTA) anticoagulant tubes were used to detect immune cells. The samples were stored at room temperature (20 ~ 30 °C) for no more than six-hour before handling. The blood sample were divided it into two tubes for flow cytometry analysis of NKT and Th1 cells respectively. Fluorescein isothiocyanate (FITC)-CD3, FITC-CD4, phycoerythrin (PE)-CD56, allophycocyanin (APC)-IFN-γ, which were purchased from eBioscience (SanDiego, CA, USA), were used for phenotypic analysis on PBMCs. The data were compensated and analyzed according to the following gating strategy: The NKT and Th1 cells were separated by setting an appropriate gate. NKT cells were doublestained with CD3-FITC and CD56-PE, and Th1 cells were doublestained with CD4-FITC and IFNG-APC. Stained cells were analyzed with BD FACSVerse flow cytometer and results were analyzed was FlowJo VX software. We performed statistical analysis and visualization of the percentage of the two cell types using GraphPad Prism 8.
Gene set enrichment analysis (GSEA)
GSEA (V4.0.3), which is a computational method that determines whether an a priori defined set of genes shows statistically significant concordant differences between two biological states [14], was performed to investigate the potential biological characteristic between high-NKT/Th1 xCell score and low-NKT/Th1 xCell score based on immune-related gene ontology gene sets downloaded from the Molecular Signatures Database [15]. We only investigated c2 (c2.cp.kegg.v7.2.symbols.gmt) gene sets in the current study. The significant cutoff value was defined as the false discovery rate (FDR < 0.25) and the normal p value < 0.05.
Statistical analysis
A student’s t-test was used to compare the means of the continuous variables between two groups. The performance of the factors and nomogram was assessed using the ROC curves (“pROC” package). A two-sided probability value less than 0.05 was considered to be statistically significant for all analyses.
Availability of data and materials
The GEO datasets (GEO ID: GSE73754) generated and/or analyzed during the current study are available in the GEO database (www. ncbi.nlm.nih.gov/gds). And all data and materials for this study shall be availed whenever requested by the editorial team, reviewers, and other users. The data set can be accessed by sending a request to 15,676,790,870@163.com.
Abbreviations
- TME:
-
Tumor microenvironment
- TIICs:
-
Tumor-infiltrating immune cell
- GSEA:
-
Gene set enrichment analysis
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- DEGs:
-
Differentially expressed genes
- AS:
-
Ankylosing spondylitis
- IL2RB:
-
Interleukin 2 receptor subunit beta
- NKT:
-
Natural killer T cells
- STAT3:
-
Signal transducer and activator of transcription 3
- SPI1:
-
Spi-1 proto-oncogene
References
Kanwal A, Fazal S. Construction and analysis of protein-protein interaction network correlated with ankylosing spondylitis. Gene. 2018;638:41–51. https://doi.org/10.1016/j.gene.2017.09.049.
Fiorillo MT, Haroon N, Ciccia F, Breban M. Editorial: ankylosing spondylitis and related immune-mediated disorders. Front Immunol. 2019;10:1232. https://doi.org/10.3389/fimmu.2019.01232.
Wu Q, Cao F, Tao J, Li X, Zheng SG, Pan HF. Pentraxin 3: a promising therapeutic target for autoimmune diseases. Autoimmun Rev. 2020;19(12):102584. https://doi.org/10.1016/j.autrev.2020.102584.
Zhang X, Lu J, Pan Z, Ma Y, Liu R, Yang S, Yang S, Dong J, Shi X, Xu S, Pan F. DNA methylation and transcriptome signature of the IL12B gene in ankylosing spondylitis. Int Immunopharmacol. 2019;71:109–14. https://doi.org/10.1016/j.intimp.2019.03.026.
Selmi C. Autoimmunity in 2018. Clin Rev Allergy Immunol. 2019;56(3):375–84. https://doi.org/10.1007/s12016-019-08745-w.
Ermoza K, Glatigny S, Jah N, Camilo V, Mambu Mambueni H, Araujo LM, Chiocchia G, Breban M. Tolerogenic XCR1(+) dendritic cell population is dysregulated in HLA-B27 transgenic rat model of spondyloarthritis. Arthritis Res Ther. 2019;21(1):46. https://doi.org/10.1186/s13075-019-1827-9.
Dulic S, Vasarhelyi Z, Bajnok A, Szalay B, Toldi G, Kovacs L, Balog A. The impact of anti-TNF therapy on CD4+ and CD8+ cell subsets in ankylosing spondylitis. Pathobiol J Immunopathol Mol Cell Biol. 2018;85(3):201–10. https://doi.org/10.1159/000484250.
Müller B, Gimsa U, Mitchison NA, Radbruch A, Sieper J, Yin Z. Modulating the Th1/Th2 balance in inflammatory arthritis. Springer Semin Immunopathol. 1998;20(1–2):181–96. https://doi.org/10.1007/bf00832006.
Min HK, Choi J, Lee SY, Seo HB, Jung K, Na HS, Ryu JG, Kwok SK, Cho ML, Park SH. Protein inhibitor of activated STAT3 reduces peripheral arthritis and gut inflammation and regulates the Th17/Treg cell imbalance via STAT3 signaling in a mouse model of spondyloarthritis. J Transl Med. 2019;17(1):18. https://doi.org/10.1186/s12967-019-1774-x.
Li X, Wang J, Zhan Z, Li S, Zheng Z, Wang T, Zhang K, Pan H, Li Z, Zhang N, Liu H. Inflammation intensity-dependent expression of osteoinductive wnt proteins is critical for ectopic new bone formation in ankylosing spondylitis. Arthritis Rheumatol. 2018;70(7):1056–70. https://doi.org/10.1002/art.40468.
Lories RJ, Haroon N. Bone formation in axial spondyloarthritis. Best Pract Res Clin Rheumatol. 2014;28(5):765–77. https://doi.org/10.1016/j.berh.2014.10.008.
Tkaczuk J, Yu CL, Baksh S, Milford EL, Carpenter CB, Burakoff SJ, McKay DB. Effect of anti-IL-2Ralpha antibody on IL-2-induced Jak/STAT signaling. Am J Transplant. 2002;2(1):31–40. https://doi.org/10.1034/j.1600-6143.2002.020107.x.
van der Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum. 1984;27(4):361–8. https://doi.org/10.1002/art.1780270401.
Tang G, Yin W. Development of an immune infiltration-related prognostic scoring system based on the genomic landscape analysis of glioblastoma multiforme. Front Oncol. 2020;10:154. https://doi.org/10.3389/fonc.2020.00154.
Lin K, Huang J, Luo H, Luo C, Zhu X, Bu F, Xiao H, Xiao L, Zhu Z. Development of a prognostic index and screening of potential biomarkers based on immunogenomic landscape analysis of colorectal cancer. Aging. 2020;12(7):5832–57. https://doi.org/10.18632/aging.102979.
Lee YH, Song GG. Meta-analysis of differentially expressed genes in ankylosing spondylitis. Genet Mol Res GMR. 2015;14(2):5161–70. https://doi.org/10.4238/2015.May.18.6.
Yang L, Wang L, Wang X, Xian CJ, Lu H. A possible role of intestinal microbiota in the pathogenesis of ankylosing spondylitis. Int J Mol Sci. 2016. https://doi.org/10.3390/ijms17122126.
Danoy P, Pryce K, Hadler J, Bradbury LA, Farrar C, Pointon J, Ward M, Weisman M, Reveille JD, Wordsworth BP, Stone MA, Maksymowych WP, Rahman P, Gladman D, Inman RD, Brown MA. Association of variants at 1q32 and STAT3 with ankylosing spondylitis suggests genetic overlap with Crohn’s disease. PLoS Genet. 2010;6(12):e1001195. https://doi.org/10.1371/journal.pgen.1001195.
Vegvari A, Szabo Z, Szanto S, Nesterovitch AB, Mikecz K, Glant TT, Adarichev VA. Two major interacting chromosome loci control disease susceptibility in murine model of spondyloarthropathy. J Immunol (Baltimore, Md: 1950). 2005; 175(4):2475–2483. https://doi.org/10.4049/jimmunol.175.4.2475
Gaafar NAG, Aslani M, Aghazadeh Z, Mortazavi-Jahromi SS, Razavi A, Mirshafiey A. Effects of mannuronic acid (M2000) on gene expression profile of signal transducer and activator of transcription proteins (STATs) in rheumatoid arthritis patients. Reumatismo. 2020;72(2):93–102. https://doi.org/10.4081/reumatismo.2020.1235.
Sims NA. The JAK1/STAT3/SOCS3 axis in bone development, physiology, and pathology. Exp Mol Med. 2020;52(8):1185–97. https://doi.org/10.1038/s12276-020-0445-6.
Zhang Y, Li X, Zhang J, Liang H. Natural killer T cell cytotoxic activity in cervical cancer is facilitated by the LINC00240/microRNA-124-3p/STAT3/MICA axis. Cancer Lett. 2020;474:63–73. https://doi.org/10.1016/j.canlet.2019.12.038.
Li J, Cao C, Xiang Y, Hong Z, He D, Zhong H, Liu Y, Wu Y, Zheng X, Yin H, Zhou J, Xie H, Huang X. TLT2 suppresses Th1 response by promoting IL-6 production in monocyte through JAK/STAT3 signal pathway in tuberculosis. Front Immunol. 2020;11:2031. https://doi.org/10.3389/fimmu.2020.02031.
Zhu F, Qiu T, Zhu S, Zhao K, Chen C, Qiao J, Pan B, Yan Z, Chen W, Liu Q, Wu Q, Cao J, Sang W, Zeng L, Sun H, Li Z, Xu K. TIRC7 inhibits Th1 cells by upregulating the expression of CTLA4 and STAT3 in mice with acute graftversushost disease. Oncol Rep. 2020;44(1):43–54. https://doi.org/10.3892/or.2020.7588.
Naruo M, Negishi Y, Okuda T, Katsuyama M, Okazaki K, Morita R. Alcohol consumption induces murine osteoporosis by downregulation of natural killer T-like cell activity. Immun Inflamm Dis. 2021. https://doi.org/10.1002/iid3.485.
Li J, Yu TT, Yan HC, Qiao YQ, Wang LC, Zhang T, Li Q, Zhou YH, Liu DW. T cells participate in bone remodeling during the rapid palatal expansion. FASEB J Off Publ Fed Am Soc Exp Biol. 2020;34(11):15327–37. https://doi.org/10.1096/fj.202001078R.
Palmer AD, Slauch JM. Envelope stress and regulation of the salmonella pathogenicity Island 1 type III secretion system. J Bacteriol. 2020; 202(17). https://doi.org/10.1128/JB.00272-20
Nguyen VC, Ray D, Gross MS, de Tand MF, Frezal J, Moreau-Gachelin F. Localization of the human oncogene SPI1 on chromosome 11, region p11.22. Hum Genet. 1990; 84(6):542–546. https://doi.org/10.1007/BF00210807
Kim K, Golubeva YA, Vanderpool CK, Slauch JM. Oxygen-dependent regulation of SPI1 type three secretion system by small RNAs in Salmonella enterica serovar Typhimurium. Mol Microbiol. 2019;111(3):570–87. https://doi.org/10.1111/mmi.14174.
Lin D, Rao CV, Slauch JM. The Salmonella SPI1 type three secretion system responds to periplasmic disulfide bond status via the flagellar apparatus and the RcsCDB system. J Bacteriol. 2008;190(1):87–97. https://doi.org/10.1128/JB.01323-07.
Hegde S, Hankey P, Paulson RF. Self-renewal of leukemia stem cells in Friend virus-induced erythroleukemia requires proviral insertional activation of Spi1 and hedgehog signaling but not mutation of p53. Stem Cells. 2012;30(2):121–30. https://doi.org/10.1002/stem.781.
Kayali S, Giraud G, Morlé F, Guyot B. Spi-1, Fli-1 and Fli-3 (miR-17-92) oncogenes contribute to a single oncogenic network controlling cell proliferation in friend erythroleukemia. PLoS ONE. 2012;7(10):e46799. https://doi.org/10.1371/journal.pone.0046799.
Yu S, Yerges-Armstrong LM, Chu Y, Zmuda JM, Zhang Y. Transcriptional regulation of frizzled-1 in human osteoblasts by Sp1. PLoS ONE. 2016;11(10):e0163277. https://doi.org/10.1371/journal.pone.0163277.
Xia CP, Pan T, Zhang N, Guo JR, Yang BW, Zhang D, Li J, Xu K, Meng Z, He H. Sp1 promotes dental pulp stem cell osteoblastic differentiation through regulating noggin. Mol Cell Probes. 2020;50:101504. https://doi.org/10.1016/j.mcp.2019.101504.
Zhang X, Li R, Qin X, Wang L, Xiao J, Song Y, Sheng X, Guo M, Ji X. Sp1 plays an important role in vascular calcification both in vivo and in vitro. J Am Heart Assoc. 2018;7(6). https://doi.org/10.1161/JAHA.117.007555
Jules J, Li YP, Chen W. C/EBPalpha and PU.1 exhibit different responses to RANK signaling for osteoclastogenesis. Bone. 2018;107:104–14. https://doi.org/10.1016/j.bone.2017.05.009.
Izawa N, Kurotaki D, Nomura S, Fujita T, Omata Y, Yasui T, Hirose J, Matsumoto T, Saito T, Kadono Y, Okada H, Miyamoto T, Tamura T, Aburatani H, Tanaka S. Cooperation of PU.1 with IRF8 and NFATc1 defines chromatin landscapes during RANKL-induced osteoclastogenesis. J Bone Miner Res Off J Am Soc Bone Miner Res. 2019; 34(6):1143–1154. https://doi.org/10.1002/jbmr.3689
Carey HA, Hildreth BE, 3rd, Samuvel DJ, Thies KA, Rosol TJ, Toribio RE, Charles JF, Ostrowski MC, Sharma SM. Eomes partners with PU.1 and MITF to regulate transcription factors critical for osteoclast differentiation. iScience.2019;11:238–245. https://doi.org/10.1016/j.isci.2018.12.018
Liu W, Di Q, Li K, Li J, Ma N, Huang Z, Chen J, Zhang S, Zhang W, Zhang Y. The synergistic role of Pu.1 and Fms in zebrafish osteoclast-reducing osteopetrosis and possible therapeutic strategies. J Genet Genom. 2020; 47(9):535–546. https://doi.org/10.1016/j.jgg.2020.09.002
Yang PM, Lin PJ, Chen CC. CD1d induction in solid tumor cells by histone deacetylase inhibitors through inhibition of HDAC1/2 and activation of Sp1. Epigenetics. 2012;7(4):390–9. https://doi.org/10.4161/epi.19373.
Chang HC, Zhang S, Thieu VT, Slee RB, Bruns HA, Laribee RN, Klemsz MJ, Kaplan MH. PU.1 expression delineates heterogeneity in primary Th2 cells. Immunity. 2005; 22(6):693–703. https://doi.org/10.1016/j.immuni.2005.03.016
Yashiro T, Takeuchi H, Kasakura K, Nishiyama C. PU.1 regulates Ccr7 gene expression by binding to its promoter in naive CD4(+) T cells. FEBS Open Bio. 2020;10(6):1115–1121. https://doi.org/10.1002/2211-5463.12861
Chang HC, Han L, Jabeen R, Carotta S, Nutt SL, Kaplan MH. PU.1 regulates TCR expression by modulating GATA-3 activity. J Immunol (Baltimore, Md : 1950). 2009;183(8):4887–4894. https://doi.org/10.4049/jimmunol.0900363
Xu X, Gao X, Zhao X, Liao Y, Ji W, Li Q, Li J. PU.1-silenced dendritic cells induce mixed chimerism and alleviate intestinal transplant rejection in rats via a Th1 to Th2 shift. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2016;38 (1):220–228. https://doi.org/10.1159/000438623
Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A, Qian J, Dong C, Yi Q. Th9 cells represent a unique subset of CD4(+) T cells endowed with the ability to eradicate advanced tumors. Cancer Cell. 2018;33(6):1048–1060. https://doi.org/10.1016/j.ccell.2018.05.004
Acknowledgements
We are grateful to Dr. Xinli Zhan (Spine and Osteopathy Ward, The First Affiliated Hospital of Guangxi Medical University,) for his generous assistance in all stages of the present study.
Funding
This work was sponsored by the National Natural Science Foundation of China (81560359), National Natural Science Foundation of China (81860393). Funding bodies had no role in the study design, collection, analysis, and interpretation of the data or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
ZX, LT: Conceptualization, Methodology. CJ, XGY, ZZ: Data curation, Investigation. XJ, ZH, JJ: Formal analysis, Software. QZ, HL, CT, YZ, NY: Visualization. LT, LC: Writing- Reviewing and Editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Ethics Committee approved this study of the First Affiliated Hospital of Guangxi Medical University. All experiments were performed in accordance with the relevant guidelines and regulations. In addition, written informed consent was obtained from each subject.
Consent for publication
All authors listed approved the publication of the manuscript.
Competing interests
We declare that we do not have any competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Cell types in the microenvironment of AS. (A, B, C, D, E) xCell score of sixty-four cell types in GSE73754 were grouped into five groups: lymphoid, myeloid, stem, stromal, and other cells.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liang, T., Chen, J., Xu, G. et al. STAT3 and SPI1, may lead to the immune system dysregulation and heterotopic ossification in ankylosing spondylitis. BMC Immunol 23, 3 (2022). https://doi.org/10.1186/s12865-022-00476-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12865-022-00476-6