
Abstract
Background
Cancer cells subvert natural immunosuppression by upregulating the expression of checkpoint proteins and their ligands. For example, tumor cells expressing programmed death-ligand 1 (PD-L1) induce immune cell tolerance to cancers, thereby facilitating tumor progression. The recent clinical success of immunotherapy, particularly checkpoint blockade, represents a significant advance in cancer therapy. However, many cancers develop resistance to immunotherapies, and the underlying mechanisms and how these might be exploited to overcome resistance still need to be determined.
Methods
T cell dysfunction, in part caused by chronic T cell receptor stimulation, diminishes the capacity for durable responses to checkpoint blockade. Furthermore, T cell populations are phenotypically and functionally heterogeneous, resulting in varying responses to checkpoint blockade. Recent molecular studies of T cell heterogeneity have shown that checkpoint blockade on its own does not alter the epigenetic landscape of T cells, despite epigenetic changes governing T cell phenotype.
Conclusion
Here we argue that epigenetic modifiers can be used to prime and sensitize T cells to immunotherapy. Administering epitherapy in conjunction with checkpoint blockade could decrease T cell exhaustion and immunotherapy resistance in many cancer types.
Similar content being viewed by others


Background
The excitement surrounding immunotherapy is justified by the positive results seen in both the preclinical and clinical settings. Advances in immunotherapy in the 19th and early twentieth century were minimal, sporadic, and overshadowed by the successes of chemo- and radiotherapy [1, 2]. However, recent immunotherapeutic discoveries such as checkpoint blockade have revolutionized cancer treatment [1, 3]. Instead of attacking the tumor directly, immunotherapies stimulate the immune system to participate in immunosurveillance; that is, recognize, mark, and destroy cancer cells [2]. An immune response against tumors is mounted in multiple stages: recognition of tumor antigens; T cell activation and proliferation; tumor infiltration with T cells; T cell killing activity; and modulation of the natural immunosuppressive response [3].
Natural immunosuppression exists to prevent autoimmunity, but it is also inadvertently detrimental to T cell efficacy. Tumor cells mediate immunosuppression by hijacking inhibitory checkpoint proteins such as programmed death 1 (PD-1), T cell immunoglobulin and mucin domain 3 (TIM-3), lymphocyte activation gene 3 (LAG-3), and cytotoxic T lymphocyte antigen 4 (CTLA-4) expressed at the surface of T lymphocytes [4, 5]. PD-1 and CTLA-4 have so far received the most attention and as such their roles in cancer immunology are now well characterized [6]. CTLA-4 and PD-1 bind specific ligands, CD80 and CD86 and PD-L1 and PD-L2, respectively, to negatively regulate and thus halt effector T cell proliferation, differentiation, and activation [3, 7]. While programmed death-ligand 1 (PD-L1) is conventionally expressed by immune cells including T cells, macrophages, and dendritic cells, tumor cells can also express PD-L1 on their cell surface and, as it confers tumors with a survival advantage.
Tumor cells exploit such immunosuppressive techniques to avoid detection by the immune system, and without immune evasion cancer could not progress from a single tumor cell to metastatic disease [2, 7]. Downstream effects of immune evasion include inhibiting T cell proliferation, inhibiting the production of cell signaling molecules (such as IL-2), and inducing T cell apoptosis [2]. Drug-induced interference of cancer cell ligands such as PD-L1 and regulatory T cell receptors makes it possible to re-instate the anti-tumor effects of T cells [8]. The first monoclonal antibodies that were tested clinically in human cancers targeted CTLA-4 and PD-1 and have now become common therapies in several cancers. Their use, termed checkpoint blockade, marked a breakthrough in cancer immunotherapy [9, 10]. The CTLA-4-blocking antibody ipilimumab entered into clinical trials in 2000 and was approved by the US Food and Drug Administration (FDA) in 2011 [9]. A 2015 study compiled data from 12 ipilimumab clinical trials and showed that ipilimumab treatment resulted in an ~ 20% five-year survival rate in patients with advanced melanoma [11] compared to the historic 10% five-year survival rate [12]. Several monoclonal antibodies have now been approved for cancer therapy by the FDA either as monotherapies or in combination with chemotherapy.
Resistance to checkpoint blockade is a major limitation of immunotherapy
While checkpoint inhibitors have no doubt been clinically successful, the problem of blockade resistance has emerged. Efforts are now underway to elucidate the mechanisms behind immune resistance to facilitate the development of therapies to overcome it [3]. There are two broad categories of immunotherapy resistance: primary resistance, which describes a tumor completely unresponsive to a novel immunotherapy; and acquired resistance, which describes tumors that initially respond to immunotherapy but then relapse and progress [3]. A third type of resistance is also sometimes referred to, termed adaptive resistance, which refers to initial recognition of the cancer by the immune system followed by the cancer selecting for immune-resistant phenotypes [2]. Given that immunotherapy use is increasing and broadening, so is acquired resistance: 25–33% of metastatic melanoma patients who initially respond to PD-1 checkpoint blockade stop responding and relapse [2].
The multifaceted, multi-step process of tumor immune responses provides many opportunities for tumors to develop immune resistance. For example, primary resistance to anti-CTLA-4 therapy can be caused by the inability of the major histocompatibility complex (MHC) to recognize tumor-associated antigens. Snyder et al. [13] discovered a peptide sequence that, when absent in melanomas, is associated with ipilimumab or tremelimumab resistance. Furthermore, there is a correlation between a high prevalence of somatic mutations (termed ‘high mutational burden’) and probability of response to checkpoint blockade, especially in melanoma [13], non-small cell lung carcinoma (NSCLC) [14], and triple-negative breast cancer (TNBC) [15]. This phenomenon occurs because a high mutational burden tumor expresses more neoantigens; that is, cancer antigens that can be recognized, marked, and attacked by the immune system [16]. PD-L1-negative tumors, for example, are associated with a low mutational burden in melanoma, primary resistance to pembrolizumab, and decreased survival [3, 17]. Snyder et al. [13] also showed that a low mutational load in melanoma leads to primary ipilimumab resistance due to a related lower affinity of tumor antigens to T cell MHC class I molecules.
Primary immunotherapy resistance can also arise from epigenetic modifications, which are heritable but reversible changes in gene expression caused by proteins bound to double helical DNA. Many of these chromatin-remodeling proteins are mutated in human cancers [18], suggesting that cancer cells exploit epigenetic patterns for tumor development and progression [19]. For this reason, the cancer epigenome has become a major therapeutic research focus, paving the way for novel cancer treatments [18, 20]. With regards to immunotherapy, tumor expression of the CXCL9 chemokine is epigenetically silenced in ovarian cancer in mice, inhibiting T cell infiltration into tumors [21]. Peng et al. [21] reversed this repression with epigenetic modifiers, resulting in re-expression of CXCL9 and an increase in T cell trafficking into tumors. Additionally, this epigenetic therapy improved tumor responsiveness to PD-L1 blockade. This is especially exciting, because ovarian cancers usually do not respond well to immunotherapy due to their low mutational burden [16]. As well as low mutational burden, “cold” tumors – which have little immune cell infiltration, such as in pancreatic and prostate cancers – also indicate poor tumor responses to checkpoint blockade [16].
In addition to the major problem of resistance, immunotherapy, like almost all drugs, has side effects. Immune-related adverse events target several systems and organs, and commonly involve the gastrointestinal tract, endocrine glands, and the respiratory, musculoskeletal and integumentary systems. As a result of the global nature of immune-related adverse events, management of these side effects warrants a multidisciplinary approach [22]. For example, the CTLA-4 antibodies ipilimumab and tremelimumab can result in immune-related adverse events that requires administration of corticosteroids and, less frequently lifelong hormone replacement therapy following inflammation of the thyroid, pituitary, and adrenal glands [9].
Finally, while checkpoint inhibition is a promising cancer treatment, many patients fail to respond or maintain a durable response, in part due to a failure to reinvigorate T cells long term or establish immunological memory in T cells [23].
T cell dysfunction
After administration of immunotherapy, T cells may successfully activate, proliferate, and infiltrate the tumor microenvironment but not perform their killing activity [3]. T cell dysfunction refers to cytotoxic T cells in the tumor microenvironment that have become ineffective or immuno-tolerant, thereby conferring both primary and acquired resistance. T cell dysfunction is in some part induced by chronic T cell receptor stimulation and therefore occurs in disease states such as chronic viral infection and cancer [24, 25]. Prolonged signaling to T cell receptors increases the expression of inhibitory immune checkpoint receptors, which in turn drive T cell dysfunction [24, 26, 27]; persistent PD-1 expression is a typical offender in that regard [9, 24,25,26,27,28,29,30,31]. Moreover, simultaneous expression of inhibitory receptors such as PD-1, CTLA-4, and TIM-3 correlates with increased T cell dysfunction in cancer [24, 28, 29] and tumor progression [27]. As the proportion of T cells that co-express these receptors increases, so does the level of dysfunction [24]. Furthermore, as the number of receptors on tumor-infiltrating lymphocytes increases, activator and effector functions decrease and tumors progress [27].
Although T cell dysfunction was initially described in lymphocytic choriomeningitis virus (LCMV) infected mice, this data laid the framework for understanding T cell dysfunction in cancer. The molecular signature of CD8+ cells in chronic viral infection has since been determined [24, 29]. Cytotoxic T cells are the pathogen killing cells of the immune system recognizable by their expression of the cell surface marker CD8. By comparing the gene expression profiles of functional and dysfunctional CD8+ cells, Wherry et al. [29] uncovered a phenotypic difference between the two states. Ribosomal subunits responsible for protein production were transcriptionally downregulated in dysfunctional CD8+ cells, perhaps explaining the subsequent reduction in cytokine production [24, 29]. Metabolic changes may be a cause of both substantial reduction in cell size and impaired proliferative ability [29]. Dysfunction progresses over time in stages, with IL-2 (a lymphocyte regulator) production diminishing early and IFNγ and TNFα loss occurring later [27]. Furthermore, T cells progress into dysfunction at various times, giving rise to considerable heterogeneity [27].
T cell exhaustion
Exhausted T cells are a distinct subset of dysfunctional T cells with poor effector function that arise in response to chronic viral infection and cancer [31, 32]. In an acute infection, effector T cells usually give rise to memory T cells but, in chronic infection and cancer, the persistent T cell receptor stimulus gives rise to progressive dysfunction until T cells become exhausted [33]. T cell exhaustion is characterized by the sustained upregulation of checkpoint proteins including PD-1, TIM-3, CTLA-4, and LAG-3 [24, 25, 29, 33]. Exhausted T cell populations are heterogeneous in function and phenotype according to varying expression of checkpoint proteins [25, 34]. Im and colleagues [34] discovered that PD-1 checkpoint blockade in LCMV only causes a proliferative response in a subset of stem-like CD8+ cells distinct from CD8+ terminal effectors, which differentiate into terminally exhausted CD8+ cells. These CD8+ cells were positive for CXCR5, the chemokine receptor, intermediate for PD-1, and negative for TIM-3, consistent with other literature [23, 35]. This population has since become known as stem-like or progenitor exhausted T cells, which are distinct from terminally exhausted tumor-infiltrating lymphocytes [25].
Several studies have now shown that checkpoint blockade can at least partially reinvigorate T cell function [33]. For example, while PD-1 blockade in NSCLC patients was successful, only a small proportion of T cell function was restored. When a high proportion of T cells had high PD-1 expression, there was minimal T cell reinvigoration, suggesting that PD-1 expression in tumor-infiltrating lymphocytes may predict responses to PD-1 blockade [27] and that the level of inhibitory receptor expressed by T cells regulates T cell functionality [33]. Conversely, terminally exhausted T cells express high levels of inhibitory receptors such as PD-1 and TIM-3 and do not respond to PD-1 checkpoint blockade in chronic viral infections [34, 35]; they do, however, have superior cytotoxicity but a shorter life span. To address why checkpoint blockade does not universally re-establish T cell effector function, Miller and colleagues [25] examined the features of specific exhausted CD8+ cell subsets. They found that dysfunctional T cell populations are heterogeneous and are composed of progenitor or stem-like exhausted T cells as well as terminally exhausted T cells. Furthermore, they found that the exhausted T cell populations in chronic viral infection and tumors have similar protein expression, whereas the exhausted population in fact comprised four major cell types: proliferating cells, effector-like cells, terminally exhausted cells, and progenitor cells.
By examining these differences between progenitor and terminally exhausted T cells, Miller et al. [25] further ascertained the functions of both populations and what facilitates or blocks responses to checkpoint blockade. Progenitor exhausted T cells were found to have superior proliferation potential and be able to differentiate into terminally exhausted cells in vivo, both contributing to their better control of tumor growth than terminally exhausted T cells. Further, PD-1 checkpoint blockade caused proliferation of progenitor exhausted cells and promoted differentiation into terminally exhausted T cells in murine melanomas [25]. PjuiTherefore, PD-1 checkpoint blockade caused temporary reinvigoration of a subset of T cells but not long-term tumor or viral control. Pauken et al. [23] found that PD-1 pathway blockade was not durable over the long term and eventually resulted in T cell “re-exhaustion”. Finally, in addition to altered protein expression in exhausted T cells, transcriptional programs are similarly altered in exhausted T cells [29, 35].
Since chronic activation promotes T cell exhaustion, it remains unclear how checkpoint blockade mediates proliferative responses of tumour-infiltrating T cells. Recently, pre-curser exhausted T cells expressing the transcription factor, TCF7, have been identified as promoters of checkpoint blockade therapy response. Transcriptome profiling of immune cells from melanoma patients identified the transcription factor TCF7 in CD8+ T cell subsets associated with positive clinical outcome of checkpoint treated patients [36]. Furthermore, Siddiqui et al. (2019) identified that intratumoral Tcf1+PD-1+CD8+ stem-like T cells are critical for tumour control in response to immunotherapy [37].
Epigenetics
As noted above, most cancers express epigenetic mutations that contribute to cancer development. Gene expression is regulated by epigenetic mechanisms that control the flux between euchromatin (open chromatin) and heterochromatin (closed chromatin). The main epigenetic mechanisms include post-translational histone modifications and DNA methylation. Post-translational histone modifications are coordinated by epigenetic enzymes that catalyze the addition and removal of functional groups. This mechanism begins with disrupted nucleosome stability, which disturbs the interactions between nucleosomes and DNA, in turn loosening or tightening higher-order chromatin folding and the switching on and off of genes and gene translation [18] (Fig. 1). For example, acetylation of a histone lysine residue neutralizes its basic nature, resulting in an overall euchromatic (open chromatin) effect [38]. Acetyl and other histone marks are covalently added by writer enzymes, and the plasticity of the epigenome is underpinned by the ability of these marks to be removed by eraser enzymes [18].

Epigenetic mechanisms such as acetylation of histones via histone acetyl transferase enzymes cause flux between euchromatin and heterochromatin. This results in altered gene expression. It is important to note that not all epigenetic enzymes are characterized as either activators or inactivators of transcription. For example, LSD1 can act as both as co-repressor or co-activator of transcription, dependent on the target residue. Cancer genomes utilize epigenetic mechanisms in tumorigenesis
Epigenetics drugs are pharmaceuticals that target proteins and genes found in the epigenome of the immune system, cancers and other diseases. Epigenetic drugs are divided into two major classes: those that target writers and those that target erasers [8, 39] . These epigenetic drugs, or epitherapies, induce anti-cancer responses by amplifying or reactivating transcription of tumor suppressor genes or cell cycle regulation genes, respectively [20]. This has downstream phenotypic effects on tumor cells including apoptosis, growth inhibition, and inducing cellular differentiation [8]. Whereas chemotherapy and radiotherapy are broad-spectrum cytotoxic treatments with side effects including autoimmune-like attack, epigenetic drugs are more targeted, with some designed to specifically identify and target proteins found only in cancer cells [39]. Several drugs targeting epigenetic witers and erasers and been FDA-approved in cancer treatment (Table 1).
Epigenetic inhibitors are able to reimburse or overcome immune resistance to immunotherapy treatment by upregulation of chemokine expression, antigen processing and presentation machinery, and immune checkpoint molecules. In particular, DNA methyltransferase (DNMT) and histone deaceytlase (HDAC) inhibitors are most well studied classes of epigenetic drugs to date. These inhibitors are able to enhance the expression of several antigen presenting molecules, co-stimulatory molecules, and checkpoint ligands [8]. Other epigenetics enzymes, such as the histone methyltransferase, EZH2, have been shown to regulate the expression of PD-L1 in hepatocellular carcinoma by upregulating H3K27me3 levels on the promoters of CD274 (encoding PD-L1) and IRF1 [40]. Furthermore, BET bromodomain inhibitors have been shown to upregulate PD-L1 and MHC I expression in ovarian and prostate cancer cells [41, 42].
Epigenetics of T cells
In addition to altered epigenetic states between tumor and normal cells, exhausted T cells are now known to have epigenetic patterns distinct from those of effector and memory T cells [23, 25, 31, 35, 43]. By comparing functional and exhausted T cells in chronic viral infection, Sen et al. [31] established that there was a large difference between their levels of chromatin accessible regions. Chromatin accessible regions were associated with increased gene expression rather than gene repression. In dysfunctional T cells, these regions were adjacent to genes overexpressed in exhausted cells such those encoding PD-1 and TIM-3, reinforcing the idea that checkpoint upregulation in dysfunctional T cells occurs at the epigenetic level and that epigenetic gene regulation programs are specific the various T cell states. Jadhav et al. [43] found two populations of T cells in LCMV mice: exhausted T cells and stem-like PD-1+ TCF1+ T cells that were epigenetically distinct. Furthermore, the stem-like T cell population was responsible for the T cell proliferative burst following anti-PD-1 therapy.
The efficacy of epitherapies in stimulating immune responses in cancer has been tested. For example, the DNA hypomethylating agent decitabine can increase PD-1 expression silenced by DNA methylation in leukemia, which, in part, makes it more responsive to PD-1 checkpoint blockade [44]. It is now suggested that DNA demethylation evokes genome wide changes, outside the PD-1 locus, which are responsible for changes in responsiveness to anti-PD-1 therapy [45].
CD8+ T cell exhaustion and T cell differentiation during exhaustion are regulated by transcription factors, particularly EOMES and T-bet [29, 46]. A study in LCMV mice found expression of genes adjacent to Eomes differed in acute versus chronic viral infection; those in acute infection were involved with effector function, whereas those in chronic infection were involved with T cell differentiation and were progressively upregulated. EOMES appears to play different roles in acute infection and T cell dysfunction [35]. PD-1high T cells are known to be associated with exhaustion, whereas PD-1int cells can be reinvigorated by checkpoint blockade. Doering et al. [46] found that T-bet was associated with different genes in PD-1high and PD-1int cells: in PD-1high cells, T-bet-associated genes included those associated with T cell exhaustion such as LAG3 and CTLA4. Therefore, PD-1 expression in heterogeneous T cell populations is driven by gene expression, consistent with the literature showing that T-bet and EOMES play a role in T cell differentiation following tumor antigen recognition.
Epitherapy and combination therapy
Therefore, many cancer patients have an inherently functional immune system that has been rendered inactive by checkpoint ligand-receptor interactions. As noted above, checkpoint blockade only partially reinvigorates the immune system and provides durable tumor immune responses in only a subset of patients [6]. Furthermore, immunotherapy has not been successful in some particularly aggressive cancers such as triple-negative breast cancer (TNBC). A clinical trial showed that only 18.5% of TNBC (so named for their lack of progesterone, estrogen, and HER-2 receptors) patients with PD-L1+ tumors responded to the anti-PD-1 antibody pembrolizumab, and the response rate to avelumab (a PD-L1 monoclonal antibody) in TNBC was only 8.6%. A clinical trial examining the efficacy of combining atezolizumab and the chemotherapy nab-paclitaxel (Abraxane) in TNBC saw a response rate of 39%; however, the duration of response was limited to nine months [47]. Another trial with 902 TNBC patients compared the administration of atezolizumab plus nab-paclitaxel with nab-paclitaxel alone, the former increasing progression-free survival from 5.5 months to 7.2 months [48].
Alternatively, epitherapy may foster an epigenetic landscape in the immune system and tumor microenvironment that is conducive to long-term immunotherapy responses (Fig. 2). Several research teams including Chiappinelli et al. [20], Jones, Issa and Baylin [18], Ribas and Wolchok [9] and Dunn and Rao [8] believe that the preclinical and clinical data strongly indicate that epitherapy is an excellent primer that could sensitize cancer cells to immunotherapy as well as minimize immunotherapy resistance. Cancer cells acquire both genetic and epigenetic mutations, and both are used to evade immune surveillance [18]. Pauken et al. [23] found that PD-1 blockade alone had little effect on the epigenetic landscape of exhausted T cells, but concluded that combining epigenetic modifying drugs with checkpoint blockade might improve the success of T cell re-invigoration. This is consistent with findings in a recent, comprehensive study by Miller and colleagues which found that PD-1 blockade had little effect on the epigenome of progenitor or terminally exhausted T cells [25]. There are currently several clinical trials underway testing the efficacy of combining epitherapy with other cancer treatments including immunotherapy, chemotherapy, and radiotherapy (Table 2).

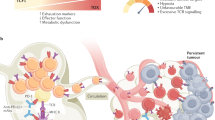
Left: As cancer cells proliferate and tumors progress, T cell receptors become chronically stimulated, lose their effector functions, and become exhausted. Middle: In contrast to immunotherapies, the administration of epitherapies alters the gene expression of T cells. Epitherapy may “level the playing field” between cancer and immune cells by reducing tumor aggression through reprogramming of cancer stem cells from the mesenchymal to the epithelial phenotype. Additionally, epitherapies prime different subsets of exhausted T cells to better respond to immunotherapy. Different classes of epigenetic therapy have distinct transcriptional roles in resetting the epigenome. Right: Once primed by epitherapies and treated with monoclonal antibodies, such as blockade to the PD-1/PD-L1 pathway, T cells have optimal capacity for reinvigoration long term
In fact, several epigenetic inhibitors, such as EZH2 and DNMT inhibitors have been shown to improve the efficacy of immunotherapy treatments such as anti-CTLA-4 and anti-PD1 treatment. For example, Goswami et al. (2018) showed that modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy in vivo [49]. Similarly, the DNMT inhibitor decitabine enhanced lymphocyte migration and function and synergized with CTLA-4 blockade in a murine ovarian cancer model [50]. Furthermore treatment with decitabine was shown to enhance the effect of PD-1 blockade in colorectal cancer by re-modulating the tumor microenvironment [51]. Improved responses have also been observed with other classes of epigenetic drugs. For example, targeted inhibition of the PD-1/PD-L1 axis by combining anti-PD-1 antibodies and the BETi JQ1 caused synergistic responses in mice bearing Myc-driven lymphomas [52]. These studies provide a strong rationale for a combination of epigenetic and immunotherapy treatment in cancer therapy.
Conclusion and future directions
Reinvigorating an ineffective immune system has become a cornerstone of cancer therapy. While monoclonal antibodies are showing great promise in promoting immunogenicity, the clinical reality is that immune reinvigoration is thwarted by primary and acquired resistance. Cancer epigenetics is an established field of significant interest in terms of both its contribution to carcinogenesis and gene expression alterations in the cancer patient’s immune system – and the complex interplay between the two. Combinations of epitherapy with established therapies have been shown to slow cancer progression at the clinical trial level, with epitherapy used to selectively reduce or re-establish the expression of genes that promote tumorigenesis and immunogenicity, respectively. Future studies in the field of epigenetics, T cell exhaustion, and cancer include developing new therapies, including combinations of therapies, for cancers unresponsive or that have low responsiveness to immunotherapy, such as prostate cancer. Furthermore, while the molecular biology of T cell exhaustion has been established, a lot of the relevant research has been in virus models and specific research into exhaustion in cancer models is warranted. Finally, many epigenetic proteins and their downstream cellular effects remain poorly characterized, even though they may have implications in cancer and T cell exhaustion. Identification of these mechanisms will facilitate further development of targeted epigenetic drugs.
Availability of data and materials
Not applicable.
Abbreviations
- AML:
-
Acute myeloid leukemia
- CLC:
-
Chronic lymphocytic leukemia;
- CRC:
-
Colorectal cancer
- CTLA-4:
-
Cytotoxic T lymphocyte antigen 4
- CXCL9:
-
CXC motif ligand 9
- DNMTi:
-
DNA methyltransferase inhibitor
- FDA:
-
US Food and Drug Administration
- HNSCC:
-
Head and neck squamous cell carcinoma
- IL-2:
-
Interleukin-2
- LAG-3:
-
Lymphocyte activation gene 3
- LCMV:
-
Lymphocytic choriomeningitis virus
- mAb:
-
Monoclonal antibody
- MHC:
-
Major histocompatibility complex
- NSCLC:
-
Non-small cell lung carcinoma
- PD-1:
-
Programmed death 1
- TCF1:
-
T cell factor 1
- TIM-3:
-
T cell immunoglobulin and mucin domain 3
- TNBC:
-
Triple-negative breast cancer
References
Mellman I, Coukos G, Glenn D. Cancer immunotherapy comes of age. Nature. 2011;480(22):480–9.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to Cancer immunotherapy. Cell. 2017;168(4):707–23.
Gide TN, Wilmott JS, Scolyer RA, Long GV. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin Cancer Res. 2018;24(6):1260–70.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Topalian S, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–61.
Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30.
Li B, Chan HL, Chen P. Immune checkpoint inhibitors: basics and challenges. Curr Med Chem. 2017;24:1–15.
Dunn J, Rao S. Epigenetics and immunotherapy: the current state of play. Mol Immunol. 2017;87:227–39.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (80- ). 2018;359(6382):1350–1355.
Couzin-Frankel J. Cancer Immunotherapy. Science (80- ). 2013;342(6165).
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of Long-term survival data from phase II and phase III trials of Ipilimumab in Unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889–94.
Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM. Systematic review of medical treatment in melanoma: current status and future prospects. Oncologist. 2011;16(1):5–24.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science (80- ). 2015;348(6230):124–128.
Katz H, Alsharedi M. Immunotherapy in triple-negative breast cancer. Med Oncol. 2018;35(1):1–9.
Vareki SM. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer. 2018;6(1):4–8.
Madore J, Strbenac D, Vilain R, Menzies AM, Yang JYH, Thompson JF, et al. PD-L1 negative status is associated with lower mutation burden, differential expression of immune-related genes, and worse survival in stage III melanoma. Clin Cancer Res. 2016;22(15):3915–23.
Jones PA, Issa JPJ, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–41.
Maio M, Covre A, Fratta E, Di Giacomo AM, Taverna P, Natali PG, et al. Molecular pathways: at the crossroads of cancer epigenetics and immunotherapy. Clin Cancer Res. 2015;21(18):4040–7.
Chiappinelli KB, Zahnow CA, Ahuja N, Bylin SB. Combining epigenetic and immunotherapy to combat cancer. Cancer Res. 2016;76(7):1683–9.
Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature [Internet]. 2015;527(7577):249–53 Available from: https://doi.org/10.1038/nature15520.
Postow MA, Sidlow R, Hellman MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378(2):158–69.
Pauken KE, Odorizzi PM, Manne S, Khan O, Chen Z, Kurachi M, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (80- ). 2016;354(6316):1160–1165.
Thommen DS, Schumacher TN. T cell dysfunction in Cancer. Cancer Cell. 2018;33(4):547–62.
Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8 + T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326–36.
Zarour HM. Reversing T-cell dysfunction and exhaustion in Cancer. Clin Cancer Res. 2016;22(8):1856–64.
Thommen DS, Schreiner J, Muller P, Herzig P, Roller A, Belousov A, et al. Progression of lung Cancer is associated with increased dysfunction of T cells defined by Coexpression of multiple inhibitory receptors. Cancer Immunol Res. 2015;3(12):1344–55.
Baitsch L, Legat A, Barba L, Marraco SA, Rivals JP, Baumgaertner P, et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS One. 2012;7(2):1–10.
Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–84.
Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–7.
Sen DR, Barnitz RA, Gerdemann U, Yates KB, Tsao H-W, Godec J, et al. The epigenetic landscape of T cell exhaustion. Science (80- ). 2016;354(6316):1165–1169.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–9.
Fuertes Marraco SA, Neubert NJ, Verdeil G, Speiser DE. Inhibitory receptors beyond T cell exhaustion. Front Immunol. 2015;6:310.
Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8 + T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537(7620):417–21.
Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37(6):1130–44.
Sade-feldman M, Yizhak K, Bjorgaard SL, Ray JP, De CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2019;175(4):998–1013.
Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1 + PD-1 + CD8 + T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity [internet]. 2019;50(1):195-211.e10. Available from: https://doi.org/10.1016/j.immuni.2018.12.021.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705.
Weintraub K. Take two: combining immunotherapy with epigenetic drugs to tackle cancer. Nat Med. 2016;22(1):8–10.
Xiao G, Jin LL, Liu CQ, Wang YC, Meng YM, Zhou ZG, et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J Immunother Cancer. 2019;7(1):1–15.
Mao W, Ghasemzadeh A, Freeman ZT, Obradovic A, Chaimowitz MG, Nirschl TR, et al. Immunogenicity of prostate cancer is augmented by BET bromodomain inhibition. J Immunother Cancer. 2019;7(1):1–14.
Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Benjamin G, Allegrezza MJ, et al. Suppressing PD-L1 expression. Cell Rep. 2017;16(11):2829–37.
Jadhav RR, Im SJ, Hu B, Hashimoto M, Li P, Lin J-X, et al. Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc Natl Acad Sci. 2019;116(28):201903520.
Yang H, Bueso-ramos C, Dinardo C, Estecio MR, Geng Q, Fang Z, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28(6):1280–8.
Ghoneim HE, Yiping F, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De novo epigenetic programs inhibit PD-1 BLOCKADEMEDIATED t-cell rejuvenation. Cell. 2017;170(1):142–57.
Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA, Michaëlsson J, et al. T-bet and Eomes Are Differentially Linked to the Exhausted Phenotype of CD8+ T Cells in HIV Infection. PLoS Pathog. 2014;10:7.
Adams S, Diamond J, Hamilton E, Pohlmann P, Tolaney S, Chang C, et al. Atezolizumab plus nab-paclitaxel in the treatment of metastatic triple-negative breast Cancer with 2-year survival follow-up: a phase 1b clinical trial. JAMA Oncol. 2018;5(3):334–42.
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast Cancer. N Engl J Med. 2018;379(22):2108–21.
Goswami S, Apostolou I, Zhang J, Skepner J, Anandhan S, Zhang X, et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J Clin Invest. 2018;128(9):3813–8.
Wang L, Amoozgar Z, Huang J, Saleh MH, Xing D, Orsulic S, et al. Decitabine enhances lymphocyte migration and function and synergizes with CTLA-4 blockade in a murine ovarian cancer model. Cancer Immunol Res. 2015;3(9):1030–41.
Yu G, Wu Y, Wang W, Xu J, Lv X, Cao X, et al. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell Mol Immunol [Internet] 2019;16(4):401–9. Available from: https://doi.org/10.1038/s41423-018-0026-y.
Hogg S, Vervoort S, Deswal S, Ott C, Li J, Cluse L, et al. BET-Bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 2017;18(9):2162–74.
Acknowledgements
I wish to thank the donors who donated funds towards the MSMTC to allow our work to continue in the field of epigenetics and cancer research.
Funding
IM is funded on an Australian Government Research Training Program Stipend Scholarship. Funds were also provided by the Melanie Swan Memorial Translational Centre (MSMTC) to cover salaries.
Author information
Authors and Affiliations
Contributions
IM and SR wrote the manuscript; WT assisted with key sections of the manuscript and aided in valuable discussions; JB created Figs. 1 and 2 with assistance from IM; JC helped generate the data for the Table 2. JD revised the original manuscript and created Table 1. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
In accordance with NHMRC guidelines and our ethical obligations as researchers, SR, WT have a financial interest in EpiAxis Therapeutics Pty Ltd. JC is also the CEO of EpiAxis Therapeutics Pty Ltd. We have in place a plan for managing any potential conflicts arising from that involvement.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
McGoverne, I., Dunn, J., Batham, J. et al. Epitherapy and immune checkpoint blockade: using epigenetic reinvigoration of exhausted and dysfunctional T cells to reimburse immunotherapy response. BMC Immunol 21, 22 (2020). https://doi.org/10.1186/s12865-020-00353-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12865-020-00353-0