
Abstract
Reliable and ultra-fast DNA and RNA sequencing have been achieved with the emergence of high-throughput sequencing technology. When combining the results of DNA and RNA sequencing for tumor cells of cancer patients, neoantigens that potentially stimulate the immune response of either CD4+ or CD8+ T cells can be identified. However, due to the abundance of somatic mutations and the high polymorphic nature of human leukocyte antigen (HLA) it is challenging to accurately predict the neoantigens. Moreover, comparing to HLA-I presented peptides, the HLA-II presented peptides are more variable in length, making the prediction of HLA-II loaded neoantigens even harder. A number of computational approaches have been proposed to address this issue but none of them considers the DNA origin of the neoantigens from the perspective of 3D genome. Here we investigate the DNA origins of the immune-positive and non-negative HLA-II neoantigens in the context of 3D genome and discovered that the chromatin 3D architecture plays an important role in more effective HLA-II neoantigen prediction. We believe that the 3D genome information will help to increase the precision of HLA-II neoantigen discovery and eventually benefit precision and personalized medicine in cancer immunotherapy.
Similar content being viewed by others

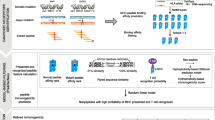

Introduction
Immunotherapies through boosting the endogenous T cell’s ability to destroying cancer cells have demonstrated effective in a variety of human malignancies [1]. The mutations of cancers can encode the seeds of their own destruction, in the form of T cell recognizable immunogenic peptides, also known as neoantigenic epitopes. There are two major origins of cancer rejection epitopes: the first origin of such antigens is formed by non-mutated proteins to which T cell tolerance is incomplete due to their restricted tissue expression pattern; the second origin is formed by peptides that are novel in normal human genome, known as neoantigens [1]. With the advance of sequencing technology, it has been revealed that during cancer development, a large number of somatic mutations can be generated. Most of these mutations are caused by genomic instability within the tumor cells and are invidious passenger mutations with unobvious growth advantage; a limited number of cancer mutations however, are driver mutations which interfere with normal cell regulation and can contribute to cancer growth and resistance to targeted therapies [2]. Both passenger mutations and driver mutations can be nonsynonymous that alter protein amino acid sequence coding, leading tumor to express abnormal proteins that cannot be found in normal cells. During cell metabolize, the proteins possessing abnormal sequences are trimmed into short peptides and are presented on the cell surface by the major histocompatibility complex (MHC, or HLA in humans) which have a chance to be recognizable by T cell as foreign antigens [2,3,4].
The cancer-killing ability of CD8+ T cells (killer T cell) can be stimulated when the T cell receptors (TCRs) recognize cancerous peptide epitopes that are displayed on major histocompatibility complex-I (MHC-I, HLA-I in human) on the surface of the tumor cells. CD4+ T cells on the other hand, identify peptides bound to MHC-II (HLA-II in human) molecules displayed on the surface of antigen-presenting cells (APCs). In recent years, CD4+ T cell (helper T cell) is drawing more attention in cancer immunotherapy area, as studies have shown that in infections or cancer, when non-self peptides or tumor-associated antigens are generated, interactions between the HLA-II–peptide complex on APCs and the TCR on CD4+ T cells, are key to initiate and sustain immune responses [5,6,7]. Comparing to HLA-I presented neoantigen discovery, due to the high polymorphic nature of HLA-II and that the HLA-II presented peptides are more variable in length, it is more challenging to efficiently predict the HLA-II loaded neoantigens [8,9,10].
According to the above principles, if candidate neoantigens are identified via sequencing experiment, one can validate the efficacy of the synthesized epitope peptides in vivo (cancer cell-line or animal model) before clinical practice [1, 2] and indeed, cancers bearing sporadic dominant mutation can often be effectively treated by targeting the driver mutation [2, 11]. When the somatic mutations are abundant however, which is a more general scenario, it is challenging to efficiently prioritize the identified neoantigen candidates according to their ability to activate the T cell’s immuno-response [12]. Although HLA peptidomics development in recent years [13,14,15] allow fast and reliable measurements of thousands of HLA ligands per sample, which improve HLA-I epitope predictions to a large extend [16,17,18,19,20], similar improvements are not hold for HLA-II, and previous studies based on high-throughput peptidomics have been restricted to a few HLA-II alleles [14, 21] or failed to demonstrate improvements in epitope predictions at all [22], leaving the space of computational HLA-II neoantigen prediction still wide open.
Over the past two decades, numerous neoantigen prediction approaches have been proposed [8, 9, 19, 23, 24] which can be partitioned into two major categories: the protein 3D structure-based approaches which consider the 3D conformations of pMHC and TCR, and the sequence-based approaches which consider the amino acid sequence of the target peptides. For the 3D structure-based approaches, if high quality pMHC 3D structures are available, molecular dynamic (MD) methods can be applied to investigate the contact affinity of pMHC-TCR complex [25,26,27], otherwise the modelling or simulation by protein docking and threading has to be employed due to the lack of high quality pMHC 3D conformation. Most other approaches adopt the sequence-based methods as there are much larger training datasets [28, 29] and the sequence-based approaches are usually more efficient to set up [12, 30].
Early sequence-based methods such as BIMAS [31] and SYFPEITHI [32] adopted the position-specific scoring matrices (PSSMs) which are defined from experimentally confirmed peptide binders of a particular MHC allele [12]. More advanced machine-learning based techniques were then developed to capture the nonlinear nature of the pMHC-TCR interaction which demonstrated better performance than the PSSM-based methods. Consensus approaches such as CONSENSUS [33] and NetMHCcons [34] that combine multiple methods were also developed to achieve more robust predictions, trading off additional computational power in determining the weighting among results generated by different methods. When considering peptide binding, most methods did not consider the HLA allele variety, therefore, pan-specific methods, such as NetMHCpan [19, 24], were developed which allow the HLA type independent prioritization. As one of the most widely adopted methods in the area, NetMHCpan first train a neural network based on multiple public datasets, then the affinity of a given peptide-MHC considering the polymorphic HLA types HLA-A, HLA-B or HLA-C is computed according to the trained neural network. NetMHCpan [19] and NetMHCIIpan [35] perform remarkably, even compared to allele-specific approaches [12, 36]. However, although several assessments and criteria were proposed in the past aiming at a more fair and effective comparison [36,37,38], there are no recent independent benchmark studies that can be used to recommend specific tools up until now [3].
In our previous study, we discovered that the DNA loci of MHC-I neoantigens obey certain distribution in genome 3D space [3] and by incorporating this important information, we developed a group feature selection based deep neural network model (DNN-GFS) that was able to predict MHC-I neoantigen in a much higher accuracy than the existing widely adopted methods [4]. To the best of our knowledge however, none of the present MHC-II epitope prediction methods consider the corresponding DNA loci of the neoantigens in the perspective of 3D genome, which carries important additional information compared to the amino acid sequence alone [39]. In this work, we incorporated the DNA origin of the immune-positive and non-negative MHC-II neoantigens in the context of the 3D genome and demonstrate its contribution to the MHC-II neoantigen prediction.
Methods
Immunogenicity data collection and curation
For MHC-II neoantigen training data, peptide sequences and the corresponding immune response information were collected from the IEDB database under the T-Cell Assay category [29] in May 2021. After collecting 399,318 peptide records in the primary dataset, we performed filtering by targeting Homo Sapiens species and MHC-II subtypes, and restrained the peptide length from 11 to 30, followed by identical records (i.e., same peptide sequence and HLA subtype) merging. The dataset was further cleaned up by applying two procedures, checkIllegalPeptides and modifyHLAType, which checks for amino-acid alphabet legitimacy and standardizes HLA allele names, respectively. For peptides of unknown MHC subtype, DRB1*01:01 was set as default MHC allele. For peptides of known MHC subtypes, we sorted them into different datasets for separate training and evaluation. Identical peptides with multiple immune experiments are defined as immuno-positive or immuno-negative if the positive rate > 70% or the positive rate < 30%, respectively. In the end, we obtained 3,633 peptides, with 2,197 immuno-negative and 1,436 immuno-positive. As for the sub data set which contains detailed MHC-II subtype information, there were in total 703 peptides, of which 411 are immuno-positive ones and 292 are immuno-negative ones.
Mapping peptides to human genome
We developed a pipeline to map the peptides sequence to reference human genome hg19; the pipeline query the NCBI local BLAST [40] and map the gene names to chromosomes and start-end positions. To set up local BLAST, we restricted the search to H.sapiens and set the E-value to 0.01 to find matches. After obtaining the accessions, we used the BIOMART [41] to convert the gene name to ENSEMBL ID, then we used the DAVID [42] to obtain the gene names composed with gene symbols and the chromosome positions were then obtained.
Chromatin 3D modeling
We used the contact frequency Hi-C data of the hESC and IMR90 cell lines generated by Bin Ren’s lab as the chromatin 3D conformation data source [43]. The contact frequencies and the subsequent chromatin 3D modeling are based on these population cell based Hi-C data. We developed a whole-genome 3D modeling algorithm for the human genome using molecular dynamics (MD) based approach with resolution of 500 kb (bin size) for hESC and IMR90 Hi-C data. Each bin was coarse-grained by the algorithm as one bead and intact genome was modeled as 23 polymer chains represented by bead-on-the-string structures [4]. Two factors would affect the spatial position of each bead: the chromatin connectivity that constrains sequentially neighbor beads in close spatial proximity and the chromatin activity that ensures active regions are more likely to be located close to the center of cell nucleus [4]. We estimated the chromatin activity as compartment degree that can be directly calculated from Hi-C matrix with algorithm described in previous work [44]. All the beads were assigned distances to the nuclear center and the conformation of chromatin was optimized from random initial structures using MD approach. The bias potential was applied to satisfy the distance constraints.
MHC-II neoantigen prediction method
We adopted the NetMHCIIpan method with Binding Affinity (BA) and Mass-Spectrometry Eluted Ligands (EL) training means respectively, to predict the curated peptides’ immunogenicity as baseline predictions. The predicted results were then treated as input feature along with the 3D genome coordinates and radius position values of hESC and IMR90. The 9 input features (1 of BA or EL, 6 of < x, y, z > coordinates from hESC and IMR90 3D models, and 2 of radius positions from hESC and IMR90 3D models) were then taken together to train KNN (K-Nearest Neighbor) with K = 8 after parameter tuning, SVM (Support Vector Machine) with default parameter of Gaussian kernel, and LR (Logistic Regression), under 5-fold and leave-one-out (LOO) cross validation schemes, to validate its contribution to the baseline prediction.
Results
After curation and generation of the two datasets, i.e., the 3,633-peptide one which contains both known and unknown MHC-II subtype p-MHCs, and the 703-peptide one which contains only known MHC-II subtype p-MHCs. We first run NetMHCPan-BA and NetMHCPan-EL to generate the NetMHCPan results as baseline predictions. We then incorporated 3D genome features, i.e., the < x, y, z > coordinates and the radius position of both hESC and IMR90 Hi-C cell lines, and trained KNN, SVM, and LR models to obtain final predictions under either 5-fold (100 repeats) or LOO cross validations. Figures 1 and 2 demonstrate the ROC curve comparison in 3,633-peptide dataset and 703-peptide dataset, respectively. In either Figure, NetMHCPan-BA or NetMHCPan-EL are the baseline predictions and Plus3D-KNN, Plus3D-LR and Plus3D-SVM are predictions after incorporating 3D genome features under cross validations. Figures 3 and 4 are the prediction score compassion of different methods under known positive and negative immunogenicity category, to demonstrate the discriminative power of different methods. The figures clearly demonstrate that after incorporating 3D genome information, the prediction accuracies are significantly boosted no matter what prediction method are used, indicating that 3D genome information can contribute more precise p-MHC-II neoantigen prediction to a large extend. Tables 1 and 2 demonstrated detailed prediction statistics at the cutoffs that reach the best F-measure score for each situation.

ROC curve comparison of different prediction methods applied on the 3,633-peptide dataset. a and b: NetMHCPan-BA results are adopted as baseline predictions and are compared with Plus3D-KNN, Plus3D-LR and Plus3D-SVM predictions, under 5-fold and Leave-one-out cross validations respectively. c and d: NetMHCPan-EL results are adopted as baseline predictions and are compared with Plus3D-KNN, Plus3D-LR and Plus3D-SVM predictions, under 5-fold and Leave-one-out cross validations respectively

ROC curve comparison of different prediction methods applied on the 703-peptide dataset. a and b: NetMHCPan-BA results are adopted as baseline predictions and are compared with Plus3D-KNN, Plus3D-LR and Plus3D-SVM predictions, under 5-fold and Leave-one-out cross validations respectively. c and d: NetMHCPan-EL results are adopted as baseline predictions and are compared with Plus3D-KNN, Plus3D-LR and Plus3D-SVM predictions, under 5-fold and Leave-one-out cross validations respectively

Positive and negative sample prediction score distribution comparison of different prediction methods applied on the 3,633-peptide dataset. a and b: NetMHCPan-BA results as baseline predictions are compared with Plus3D-KNN, Plus3D-LR and Plus3D-SVM predictions, under 5-fold and LOO cross validations respectively. c and d: NetMHCPan-EL results as baseline predictions are compared with Plus3D-KNN, Plus3D-LR and Plus3D-SVM predictions, under 5-fold and LOO cross validations respectively

Positive and negative sample prediction score distribution comparison of different prediction methods applied on the 703-peptide dataset. a and b: NetMHCPan-BA results as baseline predictions are compared with Plus3D-KNN, Plus3D-LR and Plus3D-SVM predictions, under 5-fold and LOO cross validations respectively. c and d: NetMHCPan-EL results as baseline predictions are compared with Plus3D-KNN, Plus3D-LR and Plus3D-SVM predictions, under 5-fold and LOO cross validations respectively
Discussion
The neoantigen therapy is a rising and promising strategy in cancer immunotherapy area, as it can be absolutely personalized and catch up with cancer evolution by updating neoantigen panel. It is computationally challenging however, to efficiently predict neoantigen candidates according to their ability of activating the T cell immuno-response, especially when the somatic mutations are abundant. Dozens of neoantigen prioritization or prediction approaches have been proposed to address this issue for either MHC-I or MHC-II presented epitopes, corresponding to CD8+ and CD4+ T cells respectively, but none of the existing approaches considers the DNA origin of the neoantigens from the perspective of 3D genome. In this work, we demonstrated that similar to our previous discovery for MHC-I and CD8+ T cell neoantigen, the 3D genome information can contribute to much more accurate MHC-II neoantigen prediction. The underlining mechanism why 3D genome is closely linked to neoantigen immunogenicity is yet to be revealed, but here we conjecture that it is the evolution of chromatin 3D conformation that positioned protein-coding DNA segments of different immunogenicity-activating power in specific locations in the 3D genome within the nucleus.
One perspective that worth further investigation is how the 3D genome contributes to neoantigen immunogenicity prediction by offering information into the evolutionary dynamics of genes and their regulatory elements within the nucleus. Better understanding of the 3D genome architecture allows researchers to identify how genetic elements, such as enhancers and promoters, have evolved to regulate the expression of genes. Evolution shapes these regulatory elements over time, enabling organisms to adapt and respond to different environmental and physiological conditions. When considering neoantigen immunogenicity prediction, the 3D genome can help identify the evolutionary changes that have occurred in regulatory elements controlling the expression of genes encoding tumor-specific neoantigens. Moreover, 3D genome sub-architectures such as A/B Compartment, TAD, Loop can also be further investigated along with neoantigen immunogenicity, for both MHC-I and MHC-II neoantigens, and better prediction models can be achieved by incorporating such information.
We believe that by incorporating the 3D genome information better, e.g., combining more advanced machine learning [45,46,47] and feature selection technologies [48,49,50], more precise neoantigen prioritization and discovery can be achieved and will eventually benefit precision medicine in cancer immunotherapy.
Data availability
Data and materials will be updated on https://yishi.sjtu.edu.cn/deepAntigen/.
Abbreviations
- MHC:
-
Major Histocompatibility Complex
- HLA:
-
Human leukocyte antigens
- TCR:
-
T-Cell Receptor
- IEDB:
-
The Immune Epitope Database
- BLAST:
-
Basic Local Alignment Search Tool
- MD:
-
Molecular Dynamic
- KNN:
-
K-Nearest Neighbor
- SVM:
-
Support Vector Machine
- LR:
-
Logistic Regression
- LOO-CV:
-
Leave One Out Cross Validation
References
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy, Science, vol. 348, no. 6230, pp. 69–74, Apr 3 2015.
Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. Apr 2017;17(4):209–22.
Shi Y, et al. A novel neoantigen discovery approach based on chromatin high order conformation. BMC Med Genom. 2020;13:27.
Shi Y et al. DeepAntigen: a novel method for neoantigen prioritization via 3D genome and deep sparse learning, Bioinformatics, vol. 36, no. 19, pp. 4894–4901, Dec 8 2020.
Khodadoust MS et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature, 543, 7647, pp. 723–7, Mar 30 2017.
Linnemann C et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4 + T cells in human melanoma, Nat Med, vol. 21, no. 1, pp. 81 – 5, Jan 2015.
Kreiter S et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer, Nature, vol. 520, no. 7549, pp. 692-6, Apr 30 2015.
Chen BB et al. Predicting HLA class II antigen presentation through integrated deep learning, (in English), Nature Biotechnology, vol. 37, no. 11, pp. 1332-+, Nov 2019.
Racle J et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat Biotechnol, 37, 11, pp. 1283–6, Nov 2019.
Moore TV, Nishimura MI. Improved MHC II epitope prediction - a step towards personalized medicine. Nat Rev Clin Oncol. Feb 2020;17(2):71–2.
O’Brien SG et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med, 348, 11, pp. 994–1004, Mar 13 2003.
Hackl H, Charoentong P, Finotello F, Trajanoski Z. Computational genomics tools for dissecting tumour-immune cell interactions. Nat Rev Genet, 17, 8, pp. 441 – 58, Jul 4 2016.
Chong C, et al. High-throughput and sensitive immunopeptidomics platform reveals profound interferongamma-mediated remodeling of the Human Leukocyte Antigen (HLA) Ligandome. Mol Cell Proteom. Mar 2018;17(3):533–48.
Ritz D, Sani E, Debiec H, Ronco P, Neri D, Fugmann T. Membranal and Blood-Soluble HLA Class II Peptidome Analyses Using Data-Dependent and Independent Acquisition, Proteomics, vol. 18, no. 12, p. e1700246, Jun 2018.
Bassani-Sternberg M, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun. Nov 21 2016;7:13404.
Bassani-Sternberg M, Gfeller D. Unsupervised HLA Peptidome Deconvolution improves ligand prediction accuracy and predicts Cooperative effects in Peptide-HLA interactions. J Immunol, 197, 6, pp. 2492–9, Sep 15 2016.
Bassani-Sternberg M et al. Aug., Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity, PLoS Comput Biol, vol. 13, no. 8, p. e1005725, 2017.
Abelin JG et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction, Immunity, vol. 46, no. 2, pp. 315–326, Feb 21 2017.
Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: improved Peptide-MHC class I Interaction predictions integrating eluted ligand and peptide binding Affinity Data. J Immunol, 199, 9, pp. 3360–8, Nov 1 2017.
Bulik-Sullivan B et al. Jan., Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification, (in English), Nature Biotechnology, vol. 37, no. 1, pp. 55-+, 2019.
Barra C et al. Footprints of antigen processing boost MHC class II natural ligand predictions, Genome Med, vol. 10, no. 1, p. 84, Nov 16 2018.
Garde C et al. Improved peptide-MHC class II interaction prediction through integration of eluted ligand and peptide affinity data, (in English), Immunogenetics, vol. 71, no. 7, pp. 445–454, Jul 2019.
Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res, 36, no. Web Server issue, pp. W509-12, Jul 1 2008.
Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets, Genome Med, vol. 8, no. 1, p. 33, Mar 30 2016.
Blevins SJ et al. How structural adaptability exists alongside HLA-A2 bias in the human alpha beta TCR repertoire. (in English) Proc Natl Acad Sci United States Am, 113, 9, pp. E1276-E1285, Mar 1 2016.
Riley TP, et al. T cell receptor cross-reactivity expanded by dramatic peptide-MHC adaptability, (in English). Nat Chem Biol. Oct 2018;14(10):934–.
Wang Y et al. How an alloreactive T-cell receptor achieves peptide and MHC specificity, (in English), Proceedings of the National Academy of Sciences of the United States of America, vol. 114, no. 24, pp. E4792-E4801, Jun 13 2017.
Zhang GL, Lin HH, Keskin DB, Reinherz EL, Brusic V. Dana-Farber repository for machine learning in immunology. J Immunol Methods, 374, no. 1–2, pp. 18–25, Nov 30 2011.
Vita R et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res, 47, no. D1, pp. D339-D343, Jan 8 2019.
Gupta SK, Jaitly T, Schmitz U, Schuler G, Wolkenhauer O, Vera J. Personalized cancer immunotherapy using Systems Medicine approaches, (in English). Brief Bioinform, 17, 3, pp. 453–67, May 2016.
Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains, J Immunol, vol. 152, no. 1, pp. 163 – 75, Jan 1 1994.
Schuler MM, Nastke MD, Stevanovikc S. SYFPEITHI: database for searching and T-cell epitope prediction. Methods Mol Biol. 2007;409:75–93.
Moutaftsi M, et al. A consensus epitope prediction approach identifies the breadth of murine TCD8+-cell responses to Vaccinia virus, (in English). Nat Biotechnol. Jul 2006;24(7):817–9.
Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions, Immunogenetics, vol. 64, no. 3, pp. 177 – 86, Mar 2012.
Karosiene E, Rasmussen M, Blicher T, Lund O, Buus S, Nielsen M. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ, Immunogenetics, vol. 65, no. 10, pp. 711 – 24, Oct 2013.
Trolle T et al. Automated benchmarking of peptide-MHC class I binding predictions, Bioinformatics, vol. 31, no. 13, pp. 2174-81, Jul 1 2015.
Peters B et al. A community resource benchmarking predictions of peptide binding to MHC-I molecules, PLoS Comput Biol, vol. 2, no. 6, p. e65, Jun 9 2006.
Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach, PLoS Comput Biol, vol. 4, no. 4, p. e1000048, Apr 4 2008.
Shi Y, Su XB, He KY, Wu BH, Zhang BY, Han ZG. Chromatin accessibility contributes to simultaneous mutations of cancer genes. Sci Rep. Oct 20 2016;6:35270.
Boratyn GM et al. BLAST: a more efficient report with usability improvements, Nucleic Acids Res, vol. 41, no. Web Server issue, pp. W29-33, Jul 2013.
Durinck S et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis, Bioinformatics, vol. 21, no. 16, pp. 3439-40, Aug 15 2005.
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources, (in eng). Nat Protoc. 2009;4(1):44–57.
Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485, 7398, pp. 376 – 80, Apr 11 2012.
Xie WJ, Meng L, Liu S, Zhang L, Cai X, Gao YQ. Structural Modeling of Chromatin Integrates Genome Features and Reveals Chromosome Folding Principle, Sci Rep, vol. 7, no. 1, p. 2818, Jun 6 2017.
Mnih V et al. Human-level control through deep reinforcement learning. Nature, 518, 7540, pp. 529 – 33, Feb 26 2015.
Yuan Y et al. DeepGene: an advanced cancer type classifier based on deep learning and somatic point mutations. BMC Bioinformatics, 17, no. Suppl 17, p. 476, Dec 23 2016.
Yuan Y et al. Cancer type prediction based on copy number aberration and chromatin 3D structure with convolutional neural networks, BMC Genomics, vol. 19, no. Suppl 6, p. 565, Aug 13 2018.
Cai ZP, Xu LZ. Using gene clustering to identify discriminatory genes with higher classification accuracy, (in English), Bibe 2006: Sixth Ieee Symposium on Bioinformatics and Bioengineering, Proceedings, pp. 235-+, 2006.
Yang K, Cai Z, Li J, Lin G. A stable gene selection in microarray data analysis. BMC Bioinformatics. Apr 27 2006;7:228.
Cai Z, Zhang T, Wan XF. A computational framework for influenza antigenic cartography. PLoS Comput Biol. Oct 7 2010;6(10):e1000949.
Acknowledgements
Not applicable.
Funding
This project was supported by the Key Research and Development Plan of the Ministry of Science and Technology (2022YFE0125300), the National Key Research and Development Program (2016YFC0906400), the Innovation Funding in Shanghai (20JC1418600 and 18JC1413100), the National Natural Science Foundation of China (82071262 and 81671326), the Natural Science Foundation of Shanghai (20ZR1427200 and 20511101900), the Shanghai Municipal Science and Technology Major Project (2017SHZDZX01), the Shanghai Key Laboratory of Psychotic Disorders (13DZ2260500), the Shanghai Leading Academic Discipline Project (B205), the Shanghai Jiao Tong University STAR Grant (YG2023ZD26, YG2022ZD024, and YG2022QN111), and Shanghai Gaofeng and Gaoyuan Project for the University Academic Program Development (Shanghai University of Sport-2023).
Author information
Authors and Affiliations
Contributions
MF, LL, KS and XS participated in the omics and computational experiments. LL, KS and XS assisted the computational experiments. MF provided the figures, tables, and drafted the manuscript. LM, ZG and YS designed chromatin modeling. DC, JW and GH provided clinical suggestions about cancer neoantigen presentation and biological insights of MHC-II. YS and GH initiated this project and supervised the whole workflow. YS and GH edited and finalized the manuscript. All the authors reviewed and proof read the manuscript and the experimental results.
Corresponding authors
Ethics declarations
About this supplement
This article has been published as part of BMC Genomics, Volume 25 Supplement S2, 2024: Selected articles from the 18th International Symposium on Bioinformatics Research and Applications (ISBRA-22). The full contents of the supplement are available at https://bmcgenomics.biomedcentral.com/articles/supplements/volume-25-supplement-2.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Feng, M., Liu, L., Su, K. et al. 3D genome contributes to MHC-II neoantigen prediction. BMC Genomics 25 (Suppl 2), 889 (2024). https://doi.org/10.1186/s12864-024-10687-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10687-3